
Abstract
Once a structural glass is formed, its relaxation time will increase exponentially with decreasing temperature. Thus, the glass has little chance of transforming into a crystal upon further cooling to zero Kelvin. However, a spontaneous transition upon cooling from amorphous to long-range ordered ferroic states has been observed experimentally in ferroelastic, ferroelectric and ferromagnetic materials. The origin for this obvious discrepancy is discussed here conceptually. We present a combined theoretical and numerical study of this phenomenon and show that the diffusive and displacive atomic processes that take place in structural glass and amorphous ferroics, respectively, lead to markedly different temperature-dependent relaxation behaviors, one being ‘colder is slower’ and the other being ‘colder is faster’.
Similar content being viewed by others

Introduction
The so-called ferroic glasses,1 including relaxor ferroelectrics,2, 3 ferromagnetic cluster spin glasses4 and the most recently proposed ferroelastic strain glasses,5 all have nanoscale heterogeneities in their corresponding ferroic order parameters (that is, the polarization, magnetization and strain, respectively) and characteristic internal ferroic dynamics that slows down with decreasing temperature, at least within a particular temperature range. By ferroic dynamics, we mean the involvement of long-range electrostatic/magnetostatic/elastic interactions and the absence of atomic diffusion. Even though the slowing down of ferroic dynamics with decreasing temperature seems similar to the relaxation dynamics in supercooled liquids and structural glasses, they are intrinsically different because ferroic phase transition is displacive. Indeed, recent experimental studies on ferroic materials find that an inverse type of dynamics widely exists in ferroic systems, that is, accelerating ferroic dynamics with decreasing temperature. For example, the spontaneous transition of relaxor to ferroelectrics was observed decades ago.6 Moreover, a so-called ‘spontaneous’ transition from a strain glass to a strain crystal (martensite) upon cooling has been recently reported and discussed in ferroelastic systems such as Ti-Ni-Fe shape memory alloys and Ni-Co-Mn-Ga systems.7, 8, 9 Despite these extensive experimental studies, the explanation for the accelerating ferroic dynamics with decreasing temperature remains unclear.
In this paper, we provide a combined theoretical and numerical study and show that the atomic processes that take place in structural glass and ferroic glass systems exhibit markedly different temperature-dependent relaxation behaviors, one being ‘colder is slower’ and the other being ‘colder is faster’.
Materials and methods
We consider a generic doped ferroelastic system prototype, and theoretically and numerically investigate its relaxation behavior upon cooling within a wide defect concentration range. The theoretical analysis is based on classical non-equilibrium thermodynamics and kinetic theory, and the details of the analysis are presented in the next section. The numerical simulation is based on the phase field method, which is built on the Landau theory of phase transitions and the phase field micro-elasticity theory.10, 11, 12 The details of the simulation are presented in the Supplementary Information. All of the simulations are carried out in two dimensions for the sake of simplicity because the temperature dependence of the ferroelastic transition kinetics (that is, whether colder is faster or colder is slower) will not change whether the simulation is carried out in two dimensions or in three dimensions. Then, the martensitic volume fraction, domain structure and heat capacity are calculated and analyzed throughout the temperature-induced transitions within a wide defect concentration range.
Results and discussion
Theoretical analysis
The overall kinetics of a diffusional phase transformation is typically characterized by a C-shaped curve on the time-temperature-transformation (TTT) diagram (for example, the red curve in Figure 1). The slowing down of the ferroic dynamics can also potentially lead to significant metastability, that is, a system ending up in a quite disordered ferroic state (A, defined as an ‘amorphous’ ferroic state, for example, the ferroic glass in Figure 1) that is relatively far from the theoretical optimum (X) (for example, long-range ordered domain structures), but the ferroic relaxation (ageing) would be sluggish when compared with the laboratory timescale such that one must reckon with A. There are discussions in the strain glass literature regarding whether the source of the ferroic disorder (also known as nanoscale heterogeneities) at lower temperatures in A is due to a dynamical arrest of the thermally equilibrated ferroic disorder of higher temperatures (strain liquid), or a quenched-in ‘diffusive’ disorder (chemical) from even higher temperatures13, 14, 15 or both. For this study, however, the exact source of this ferroic disorder is irrelevant. Here it makes no difference whether one is dealing with the ferroic analog of the liquid melt-quench-produced structural glass or the various forms of ‘solid-state’ amorphization (such as ion implantation, oxidation, stress-induced amorphization and so on) that have not experienced the characteristic liquid-to-glass transition often used in textbooks. The only hypothesis we make in our theory is that the relaxation rate at A is so slow that significant ferroic disorder survives for a period of time that affects the application of interest. An analogy is the native silicon oxide formed by air oxidation on cleaved silicon surfaces, where diffusive relaxation is so slow that the silicon oxide is not transformed to crystalline quartz.

Schematic time-temperature-transformation (TTT) curves for ferroelastic systems with three typical doping levels (mole fraction c=0.02, 0.14 and 0.40). The vertical and horizontal axes represent for temperature (T) and time (t), respectively. ‘X’ represents the theoretical optimum, that is, martensite, and ‘A’ represents the amorphous strain state in the ferroelastic system. See text for discussions of the shapes of the three TTT curves.
For the convenience of our discussion, we define the relaxation rate R (inverse of relaxation time) as

where υD~1012 s−1 is a fixed frequency for each attempt, taken as the Debye frequency, and Q(T) is an effective activation energy. To avoid controversy in our terminology, we will use the term ‘amorphous ferroics’ instead of ‘ferroic glasses’ to describe the present state A that has more ferroic disorder than X. Q(T) is a lumped parameter16 containing both the driving force (for example, the drop in the free energy when the native silicon oxide transforms to quartz in the example above) and the microscopic pathway sampling (that is, kinetics). We quantify the driving force by the following:

where F is the constrained Helmholtz free energy within the metabasin A or X in the r3N phase space (where N is the number of atoms) obtained by integrating the partition function within a metabasin:

where U(r3N) is the potential energy. The normalization of Equation (2) by volume is to make the driving force an intensive quantity. Note that Δf has the same unit as stress (MPa), and can be considered a thermodynamic driving force. We can then model the two contributions to Q(T)—the driving force dependence and the energy landscape ‘ruggedness’ dependence—more explicitly by defining a two-variable function as follows:

where the first dependency on the right-hand side is the driving force and the second dependency comes from the microscopic pathway sampling.17 To accentuate the first dependency, we define an activation volume by the following:

where ‘T in second entry’ refers to the T outside Δf that is also a function of temperature. The definitions above are general and apply to both the diffusion-dominated and displacement-dominated atomic relaxations (for an atomistic definition of diffusive/civilian vs displacive/military processes, see refs 18, 19). However, the activation volume Ω of displacement-dominated processes (such as deformation twinning) is generally much larger than that of diffusion-dominated processes (such as diffusional creep) because the latter are much more one atom’s ‘decision’ than many atoms’ collective action at the saddle point.20 Ferroic relaxations are classified as displacement-dominated processes and, according to Cross’ estimate,2 involve changes of ferroic order in an ~(10 nm)3 volume in relaxor ferroelectrics, which is equivalent to an ~104 atomic volume that is much larger than the Ω of typical atomic diffusion (usually <1 atomic volume21). Therefore, the following holds

This can have a profound consequence on the TTT behavior. In considering the example of the native oxide again, if a native oxide is amorphous at room temperature, it will not crystallize into quartz as T→0 K. In the liquid melt-quench case, once the opportunity window for crystallization (the TTT nose) is missed, the system ends up as a structural glass at any lower temperatures. We are confident about the eventual amorphous outcome in these two diffusive examples because  and, therefore, the diffusional relaxation rate R(T) in Equation (1) decrease monotonically as T→0 K. This is because as T decreases, although Q(T) may drop, it does not drop as fast as T, and T will reach zero first, giving rise to an infinitely slow relaxation (aging) at T=0 K and a permanent amorphous state. Therefore, once the disorder appears at a specific temperature, the disorder is maintained while the temperature decreases further. As we will show below, this is no longer the case when the activation volume Ω is large enough, that is, the ferroic relaxation can instead accelerate with decreasing temperature, resulting in a decrease in the ferroic disorder in A as the temperature decreases, and eventually the amorphous ferroics may crystallize into X at a lower temperature.
and, therefore, the diffusional relaxation rate R(T) in Equation (1) decrease monotonically as T→0 K. This is because as T decreases, although Q(T) may drop, it does not drop as fast as T, and T will reach zero first, giving rise to an infinitely slow relaxation (aging) at T=0 K and a permanent amorphous state. Therefore, once the disorder appears at a specific temperature, the disorder is maintained while the temperature decreases further. As we will show below, this is no longer the case when the activation volume Ω is large enough, that is, the ferroic relaxation can instead accelerate with decreasing temperature, resulting in a decrease in the ferroic disorder in A as the temperature decreases, and eventually the amorphous ferroics may crystallize into X at a lower temperature.
Consider the change in relaxation rate with temperature as follows:

where

is the entropy difference between A and X per volume. The quantity

can be defined as the effective activation enthalpy according to the Gibbs–Helmholtz relation. So we obtain the critical condition for a transition from ‘colder is slower’ to ‘colder is faster’:

which gives a particularly simple physical picture: if the excess entropy (ΔsAX) of A with respect to X within the activation volume Ω exceeds the enthalpic barrier (H) of relaxation between A and the next metabasin A′ (A→A*→A′, where A′ is the next metabasin on the path of aging and A* is the saddle point in between),20 then the change in the relaxation rate becomes positive upon cooling and the relaxation dynamics will accelerate with decreasing T (that is, colder is faster), and vice versa.
The excess entropy ΔsAX can be evaluated from first principles. In the diffusional transformation examples, if A is liquid and X is crystal, from Richard’s rule, ΔsAX would be ~kB per atomic volume. If A is ergodicity-limited to a single vibrational basin, then ΔsAX would be the vibrational entropy difference that may reasonably be ~0.1 kB per atomic volume. However, in general, because diffusional transformations have a small activation volume Ω20, the right hand side of Equation (10) is ‘activation enthalpy H-dominated’ and cannot turn negative. Thus, the system will stay in the ‘colder is slower’ regime, and the amorphous state would remain even with a further decreasing T.
However, the case for ferroic materials is quite different and is dependent on defect concentration c. Figure 2 illustrates the effective activation enthalpy H and entropy TΩΔsAX for ferroics with different values of c at a constant temperature T, which is lower than the thermodynamic equilibrium transition temperature. As c increases, the effective activation enthalpy H for the A→X transition increases (that is, it is more difficult for the A→X transition to take place) because the randomly distributed point defects pose energetic barriers that need to be overcome to form a long-range ordered ferroic state. However, the effective activation entropy TΩΔsAX has the opposite trend. On one hand, there is no huge variation in the magnitude of ΔsAX because, as mentioned above, a vibrational entropy difference of ~0.1 kB per atomic volume seems appropriate. On the other hand, the activation volume  should decrease with increasing c by a simple geometric scaling argument because more random point defects make the transition less collective. Consequently, we can divide the whole range of the concentration of point defects into two parts. The first part is before the intersection point (cc in the horizontal axis of Figure 2) of these two curves (region 1 and 2 combined): here the final phase upon cooling to T=0 K is X, and the growth of nanodomains is activation entropy-dominated, thus ‘colder is faster’; the second part is after the intersection point (region 3): here the nanodomain growth and coarsening are always activation enthalpy-dominated, thus ‘colder is slower’, such as in structural glasses (we refer to it as the ‘permanent amorphous ferroic’ (PAF) state here because the ferroic disorder can survive down to T→0 K). The temperature we consider here is lower than the corresponding nucleation temperature, thus, the nanodomains can exist steadily but the whole system is still far from X.
should decrease with increasing c by a simple geometric scaling argument because more random point defects make the transition less collective. Consequently, we can divide the whole range of the concentration of point defects into two parts. The first part is before the intersection point (cc in the horizontal axis of Figure 2) of these two curves (region 1 and 2 combined): here the final phase upon cooling to T=0 K is X, and the growth of nanodomains is activation entropy-dominated, thus ‘colder is faster’; the second part is after the intersection point (region 3): here the nanodomain growth and coarsening are always activation enthalpy-dominated, thus ‘colder is slower’, such as in structural glasses (we refer to it as the ‘permanent amorphous ferroic’ (PAF) state here because the ferroic disorder can survive down to T→0 K). The temperature we consider here is lower than the corresponding nucleation temperature, thus, the nanodomains can exist steadily but the whole system is still far from X.

Effective activation enthalpy H (black curve) and effective entropy within activation volume ΩΔsAX (red curve) change along with defect concentration at a certain temperature (schematic). Here cc represents a critical point defect concentration where ‘colder is faster’ transfers into ‘colder is slower’.
Furthermore, we can divide the former part further into two regions. Below a certain concentration (region 1), a normal first-order ferroic transition takes place. After this concentration (region 2), the parent phase will first jump into a metastable state (A) and then later escape from this state to X through a colder-is-faster nanodomain growth and coarsening. Because this metastable state is in transient disorder (it exists at intermediate temperatures but cannot survive to 0 K), we call it the ‘transient amorphous ferroic’ (TAF) state. Thus, the standard ultrafast ‘almost instantaneous’ ferroic transition takes place in region 1, the TAF and spontaneous A→X transition occur in region 2 but before the intersection point and the PAF state appears in region 3.
In summary, because of the two competing activation parameters of comparable magnitude in ferroic transitions, the right-hand side of Equation (10) may turn from ‘activation entropy-dominated’ to ‘activation enthalpy-dominated’ as we increase the concentration of point defects, and the ferroic relaxation dynamics would change from ‘colder is faster’ (leading to TAF) to ‘colder is slower’ (leading to PAF). Indeed, the plausibility of TAF hinges on the scenario where Q(T) hits zero before T reaches zero, as it is common for displacive processes (dislocation slip, fracture, deformation twinning and so on) to have the so-called ‘athermal threshold’20 Δfath:

If Δf (T0>0)=Δfath, the ferroic relaxation dynamics would become infinitely fast as T→T0 from above. In this case, the amorphous ferroics can never survive below T0 and therefore, the ferroic disorder must be in the TAF condition (as shown by the horizontal blue line in the TTT diagram in Figure 1). Such accelerating ferroic dynamics with decreasing temperature in amorphous ferroics should also have interesting internal friction signatures22 above T0. Please note that ferroic relaxation under a small driving force has been analyzed by Vugmeister et al.23 using a linear-response theory. In the case of a large driving force, however, such dependence becomes the main driver of the novel ‘colder is faster’ phenomenon discussed above and cannot be ignored.
Numerical simulation
To verify the analytical theory above, we carry out two-dimensional phase field simulations of detailed microstructural changes upon cooling in a generic ferroelastic system that undergoes a cubic to tetragonal martensitic transformation (MT). Three different concentrations of doped point defects are considered and the simulation results obtained are shown in Figure 3. When the point defect concentration c=0.02, the first figure in the top row of Figure 3 shows the microstructure of the parent phase (which will be hereafter referred to as austenite) at 281 K, where the green color represents the austenite and the deviations from the green color describe the structural nonuniformities (lattice distortions) associated with martensitic embryos induced by the doped defects. When T decreases to 217 K (TP-M, that is, the austenite to martensite transition temperature), the system shows a sudden appearance of twinned martensitic plates, with equilibrium values (red and blue) of the order parameters introduced in the phase field model to describe the two Bain variants of martensite. This can also be detected by the change in the martensitic volume fraction and its derivative with respect to temperature (Supplementary Figure S1a and b). At TP-M, the martensitic volume fraction shows a sudden jump and then gradually approaches 100%. Correspondingly, a sharp peak appears at TP-M in the derivative of the martensitic volume fraction with respect to T. In addition, the sharp peak shown in the calculated differential scanning calorimetry curve (Supplementary Figure S2) at TP-M clearly indicates an ultrafast ‘almost instantaneous’ characteristic of the standard MT. After the phase transition, the system has a well-developed long-range-order (LRO) strain pattern consisting of two different martensitic variants in a polytwin structure. The structure is stable till the end of the cooling process at 48 K (see the last figure in the top row of Figure 3). Supplementary Figure S1c and d shows the change in the number of domains upon cooling. At this low doping level (c=0.02), the domain number first increases and then decreases to a relatively small value, consistent with the final structural state of the system shown in Figure 3; this state consists of well-developed large martensitic twin domains. Thus, when the concentration is low, point defects cannot prevent the LRO twinning martensite from forming.

Microstructure evolutions upon cooling in three ferroelastic systems having different defect concentrations, c=0.02, 0.14 and 0.40, respectively. Green color in the figures represents the parent phase; blue and red colors represent the two different martensitic variants. The final state of cooling is 48 K.
When c=0.40 (the bottom row of Figure 3), the microstructure changes upon cooling are fundamentally different. There are no sudden changes in the number of domains, domain size and volume fraction of martensite. Although spatial correlations among more mature martensitic domains start to develop upon cooling, leading to tweed-like patterns,24 the LRO twinning plates never form. The increase in the total martensitic volume fraction starts at a much lower temperature (~130 K) and is rather slow (Supplementary Figure S1a) upon further cooling compared with the previous case. In addition, it reaches only ~35% rather than 100% at the end of cooling. The number of domains starts to increase at the same temperature (~130 K), with the increasing rate (the slope of the domain number vs temperature curve shown in Supplementary Figure S1c) reaching its maximum value at ~114 K and the number of domains reaching (gradually, as shown in Supplementary Figure S1b) its maximum value at ~68 K (Supplementary Figure S1a). Although the domain number starts to decline upon further cooling, more than 80% of the domains survived at the end of cooling. Thus, when the defect concentration is high, the system ends up in the PAF condition, with a nanoscale ferroic disorder capable of surviving to T→0 K25, 26, 27 that never transforms into the LRO twinning martensite. This is because ‘colder is slower’ has taken hold and T reaches zero before Q(T) does, as in the relaxation dynamics of conventional structural glass.
The middle row in Figure 3 shows the microstructure evolution in a system in the crossover region on the ferroelastic strain-state phase diagram,7 that is, the region with a defect concentration of c=0.14 in the model system considered in this study. The simulation results show a ‘diffuse’ MT, that is, the system eventually transforms into the LRO twinning martensite, but the transition occurs in a wide temperature range (~13 K), which is in sharp contrast to that of normal MT that occurs in the system with c=0.02. Above 195 K, only martensitic nanodomains exist in the systems, but the peak at temperature TP-A in Supplementary Figures S1b and S2 suggests that individual domains of martensite begin to nucleate and grow rapidly below this temperature; however, no interconnected martensitic plates are formed yet. At 182 K (TA-M), domains are interconnected and LRO twinned martensitic plates emerge. Correspondingly, the second peak appears in Supplementary Figures S1b and S2, suggesting that a transition from a TAF state (a metastable state similar to the last figure in the bottom row of Figure 3) to a normal martensitic structure by ‘displacive domain growth and coarsening’ has occurred at this temperature. With continued cooling, the internally twinned martensitic structure develops further (with a higher degree of perfection) and remains till the end. In this case, a two-step transition, austenite→TAF state→LRO twinning martensite takes place.
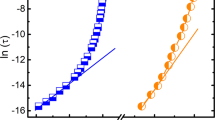
Figure 4a shows the Landau free energy curves of the system at 255 and 245 K. We can see that the activation energy Q decreases, whereas the driving force Δf increases as temperature decreases. Both of these make the MT easier. A detailed analysis of the temperature-dependence of Q/T and Δf is displayed in Figure 4b. Although Δf scales linearly with temperature (the green curve Figure 4b), Q/T is more sensitive to temperature because Q is also a strong function of Δf; thus,  , which can be readily observed from the blue curve in Figure 4b. Therefore, the MT kinetics will accelerate with decreasing temperature, making the percolation of martensitic domains and formation of LRO twinning plates (that is, spontaneous A to X transition) more observable at lower temperatures.
, which can be readily observed from the blue curve in Figure 4b. Therefore, the MT kinetics will accelerate with decreasing temperature, making the percolation of martensitic domains and formation of LRO twinning plates (that is, spontaneous A to X transition) more observable at lower temperatures.

Thermodynamics and kinetics of the ferroelastic system. (a) The free energy curves for austenite→martensite transition at 255 and 245 K, respectively. Q is the activation energy and Δf is the driving force. (b) The temperature-dependence of Q/T (blue dot line) and Δf (green dot line).
As was stated earlier, the essential difference between structural glass and amorphous ferroics lies in their TTT curves (Figure 1). There is a single-type C-shaped curve (the red curve) for the structural glass to crystal transition, where the diffusive atomic relaxation process is always ‘colder is slower’ once the nucleation difficulty is overcome, while three different types of TTT curves can be anticipated for the ferroics at three typical point defect concentration levels, as illustrated schematically in Figure 1. These TTT curves are associated with the additional kinetic difficulty of ‘displacive growth and coarsening’ that has different characteristics from diffusional processes. The dashed line is a cooling line with a constant cooling rate, and the dark blue, light blue and red curves are for the ferroelastic systems considered in the simulations with a defect concentration of 0.02, 0.14 and 0.40, respectively. When the point defect concentration is low (0.02), a first-order MT takes place at T0. This transition happens in such a short time that we can almost neglect the role of time, which means that the transition temperature almost remains constant as the cooling rate changes. In this case, the TTT curve is a horizontal line (dark blue). As the point defect concentration increases (at 0.14 for instance), the TTT curve (light blue) has a ‘half-C’ shape and the behavior of the system becomes quite different. The parent phase (strain liquid) will first transform into an amorphous ferroelastic state (that is, frozen strain disorder) at a temperature slightly lower than T0. The system remains in such an amorphous state (that is, the TAF state mentioned above) owing to the sluggish ‘displacive domain growth and coarsening’ within the temperature range (T0 and T1), and the amorphous state to martensite transition (the LRO strain state) will take place upon further cooling to temperatures below T1. The range (T0 and T1) where the TAF state exists depends on the specific cooling rate. At a higher defect concentration (for example, c=0.4), the TTT curve (red) has a normal C shape. If the cooling rate is large enough (black dashed line), the observation time t2 will be smaller than the critical time needed to observe the MT, and thus the cooling line does not intersect with the TTT curve at this doping level. Consequently, we can only observe the austenite→amorphous ferroelastic state transition, and the system will be trapped in a PAF state, with nanoscale ferroic disorder capable of surviving to T→0 K. Thus, a highly doped ferroelastic system is expected to behave similarly to a structural glass owing to the dominance of the activation enthalpy term in the relaxation kinetics.
Conclusion
In conclusion, we have formulated a theoretical framework for analyzing the temperature and defect concentration dependence of ferroic relaxations and ferroic transitions. The framework reveals the nature of the accelerating ferroic dynamics (post-nucleation displacive domain growth and coarsening) upon cooling and unveils a change from ‘colder is faster’ to ‘colder is slower’ with an increasing defect concentration. The origin of such a transition arises from the fact that there are two competing activation parameters of potentially comparable magnitude for ferroic dynamics, that is, the activation entropy and activation enthalpy, as a result of the much larger activation volumes of displacive atomic processes compared with those of diffusional processes. When the defect concentration is low, the ferroic relaxation is dominated by the activation entropy, and the relaxation is ‘colder is faster’. However, as the defect concentration increases, the activation enthalpy will become dominant and the relaxation kinetics changes to ‘colder is slower’. There are two types of ferroic disorders associated with this parametric change: one (TAF) that cannot survive to T→0 K and transforms to ferroic crystal, and one (PAF) that can survive and transforms to permanent amorphous ferroics or ferroic glasses. These theoretical predictions have been verified by phase field simulations of the microstructural (strain state) evolution during a generic ferroelastic phase transition upon cooling in systems with three different defect concentration levels.
References
Entel, P., Arroyave, R., Fähler, S., Kainuma, R., Planes, A., Ren, X. & Saxena, A. Ferroic glasses: magnetic, polar and strain glass. phys. status solidi b 251, 1965–1966 (2014).
Cross, L. E. Relaxor ferroelectrics. Ferroelectrics 76, 241–267 (1987).
Bokov, A. A. & Ye, Z. G. Recent progress in relaxor ferroelectrics with perovskite structure. J. Mater. Sci. 41, 31–52 (2006).
Sherrington, D. A spin glass perspective on ferroic glasses. phys. status solidi b 251, 1967–1981 (2014).
Sarkar, S., Ren, X. & Otsuka, K. Evidence for strain glass in the ferroelastic-martensitic system Ti 50-x Ni 50+ x. Phys. Rev. Lett. 95, 205702 (2005).
Chu, F., Reaney, I. & Setter, N. Spontaneous (zero‐field) relaxor–to–ferroelectric‐phase transition in disordered Pb (Sc1/2Nb1/2) O3. J. Appl. Phys. 77, 1671–1676 (1995).
Zhang, J., Wang, Y., Ding, X., Zhang, Z., Zhou, Y., Ren, X., Wang, D., Ji, Y., Song, M. & Otsuka, K. Spontaneous strain glass to martensite transition in a Ti 50 Ni 44.5 Fe 5.5 strain glass. Phys. Rev. B 84, 214201 (2011).
Wang, Y., Huang, C., Wu, H., Gao, J., Yang, S., Wang, D., Ding, X., Song, X. & Ren, X. Spontaneous strain glass to martensite transition in ferromagnetic Ni-Co-Mn-Ga strain glass. Appl. Phys. Lett. 102, 141909 (2013).
Ren, X. Strain glass and ferroic glass—unusual properties from glassy nano‐domains. phys. status solidi b 251, 1982–1992 (2014).
Kartha, S., Castán, T., Krumhansl, J. A. & Sethna, J. P. Spin-glass nature of tweed precursors in martensitic transformations. Phys. Rev. Lett. 67, 3630 (1991).
Semenovskaya, S. & Khachaturyan, A. Coherent structural transformations in random crystalline systems. Acta. Mater. 45, 4367–4384 (1997).
Zhou, Y., Xue, D., Ding, X., Wang, Y., Zhang, J., Zhang, Z., Wang, D., Otsuka, K., Sun, J. & Ren, X. Strain glass in doped Ti 50 (Ni 50− xDx)(D= Co, Cr, Mn) alloys: implication for the generality of strain glass in defect-containing ferroelastic systems. Acta. Mater. 58, 5433–5442 (2010).
Kustov, S., Salas, D., Cesari, E., Santamarta, R., Mari, D. & Humbeeck, J. V. Strain glass revisited. Mater. Sci. Forum 738–739, 274–275 (2013).
Kustov, S., Salas, D., Cesari, E., Santamarta, R., Mari, D. & Van Humbeeck, J. Structural anelasticity, elasticity and broken ergodicity in Ni-Ti shape memory alloys. Acta. Mater. 73, 275–286 (2014).
Liu, J. & Jin, X. Is ‘strain glass’ a nanodomain frozen transition? phys. status solidi b 251, 2055–2060 (2014).
Kushima, A., Lin, X., Li, J., Eapen, J., Mauro, J. C., Qian, X., Diep, P. & Yip, S. Computing the viscosity of supercooled liquids. J. Chem. Phys. 130, 224504 (2009).
Li, J., Kushima, A., Eapen, J., Lin, X., Qian, X., Mauro, J. C., Diep, P. & Yip, S. Computing the viscosity of supercooled liquids: Markov network model. PLoS ONE 6, e17909 (2011).
Wang, C.-C., Mao, Y.-W., Shan, Z.-W., Dao, M., Li, J., Sun, J., Ma, E. & Suresh, S. Real-time, high-resolution study of nanocrystallization and fatigue cracking in a cyclically strained metallic glass. Proc. Natl Acad. Sci. USA 110, 19725–19730 (2013).
Mao, Y., Li, J., Lo, Y.-C., Qian, X. & Ma, E. Stress-driven crystallization via shear-diffusion transformations in a metallic glass at very low temperatures. Phys. Rev. B 91, 214103 (2015).
Li, J. The mechanics and physics of defect nucleation. MRS Bull. 32, 151–159 (2007).
Zhu, T. & Li, J. Ultra-strength materials. Prog. Mater. Sci. 55, 710–757 (2010).
Hamzić, A. & Campbell, I. [The spin glass to ferromagnetic order transition in AuFe alloys]. J. Phys. Lett. 42, 309–312 (1981).
Vugmeister, B. & Rabitz, H. Dynamics of interacting clusters and dielectric response in relaxor ferroelectrics. Phys. Rev. B 57, 7581 (1998).
Kartha, S., Krumhansl, J. A., Sethna, J. P. & Wickham, L. K. Disorder-driven pretransitional tweed pattern in martensitic transformations. Phys. Rev. B 52, 803 (1995).
Zhang, Z., Wang, Y., Wang, D., Zhou, Y., Otsuka, K. & Ren, X. Phase diagram of Ti 50−x Ni 50+x: Crossover from martensite to strain glass. Phys. Rev. B 81, 224102 (2010).
Semenovskaya, S. & Khachaturyan, A. G. Coherent structural transformations in random crystalline systems. Acta Mater. 45, 18 (1997).
Kartha, S., Castan, T., Krumhansl, J. A. & Sethna, J. P. Spin-glass nature of tweed precursors in martensitic transformations. Phys. Rev. Lett. 67, 4 (1991).
Acknowledgements
This work was supported by the National Basic Research Program of China (2012CB619402 and 2014CB644003), the National Key Research and Development Program of China (2016YFB0701302), National Natural Science Foundation of China (51671156) and 111 Project of China (B06025). YW acknowledges support by the US. Natural Science Foundation (NSF) DMR-1410322. JL acknowledges support by NSF DMR-1410636.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the NPG Asia Materials website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, J., Mao, Y., Wang, D. et al. Accelerating ferroic ageing dynamics upon cooling. NPG Asia Mater 8, e319 (2016). https://doi.org/10.1038/am.2016.152
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2016.152
