
Abstract
We constructed a linkage map of Bombus terrestris (Hymenoptera, Apidae) phase unknown. The map contains 79 markers (six microsatellite and 73 RAPD markers) in 21 linkage groups and spans over 953.1 cM. The minimal recombinational size of the B. terrestris genome was estimated to be 1073 cM. Using flow cytometry, the physical size of the haploid genome of B. terrestris was calculated to be 274 Mb. This is the second linkage map for a social insect species. Bombus terrestris has on average five times less recombinational events per kb than the honey bee Apis mellifera. Male haploidy, chromosome size, and eusociality can now be excluded as reasons for the high recombination frequency of Apis mellifera. Finally, the sex determination locus of B. terrestris was placed on the map using bulked segregant analysis.
Similar content being viewed by others

Introduction
Since Dzierzon’s (1845) pioneering work on honey bees it has long been known that male Hymenoptera are haploid and develop from unfertilized eggs. This so called haplo-diploid sex determination is used by approximately 20% of all animals (Bull, 1983). Under haplo-diploidy males develop from unfertilized eggs and are haploid whereas females are diploid and arise from fertilized eggs. The most widespread proximate mechanism of sex determination in Hymenoptera seems to be the complementary sex determination system (CSD; Whiting, 1943; for reviews see Crozier, 1977 and Cook, 1993). The idea behind CSD is that individuals which are hemi- (= haploid) or homozygous at the sex locus will develop into phenotypic males whereas heterozygous individuals are females (Whiting, 1943). A particular case of CSD is the single-locus CSD (e.g. Butcher et al., 2000), i.e. there is only a single sex determination locus. If species with a single-locus CSD are inbred (e.g. a sister–brother mating) we expect that half of the diploids will be males. Those diploid males are either inviable, sterile or produce sterile (triploid) daughters. Therefore, the effects of inbreeding in Hymenoptera with a single-locus CSD are severe. Evidence for single-locus CSD is mostly provided by controlled breeding experiments and the occurrence of diploid males at the expected frequency (Cook, 1993). However, the molecular basis of both the haplo-diploidy and CSD remains unknown.
Apis mellifera (the honey bee) has a single-locus CSD and is the only social hymenopteran species for which the location of the sex determination locus is currently mapped both in a linkage and a physical map (Hunt & Page, 1994; Beye et al., 1996). Here, we extend this analysis by mapping the sex locus in the bumblebee, Bombus terrestris, in order to provide a second system to the investigation of the molecular mechanisms of sex determination under haplo-diploidy. Bumblebees belong to the same family as honeybees (Apidae). As a study system, bumblebees have the advantage that diploid males are raised to adulthood, in contrast to honey bees where they are normally removed as larvae (Woyke, 1963).
Bombus terrestris is a well-studied and widespread species in Europe, and breeding experiments suggested a single-locus CSD (Duchateau et al., 1994). On the basis of ratios seen in the neotropical species B. atratus Garofalo (1973) proposed a two-locus CSD system. Duchateau et al. (1994) on the basis of breeding studies showed clearly that a single-locus CSD system applied to B. terrestris. Although it is possible that congeners differ in sex-determination system, a sample size effect is more likely, given that Garofalo’s (1973) results do not differ significantly from those expected under one-locus CSD (Crozier, 1977). We here place the single sex determination locus of B. terrestris on a genetic map, an important step to understand the molecular mechanisms that determine sex under male haploidy in Hymenoptera. Diploid males and females (workers or gynes in the case of social Hymenoptera) derived from one queen differ under a single-locus CSD system in the allelic composition at the sex locus; diploid males have to be homozygous whereas workers have to be heterozygous. This can be used to map the sex determination locus because molecular markers that are linked with the sex locus should show the same pattern.
One recurrent problem for the mapping of social Hymenoptera is that only a few species can be bred in the laboratory, where control over ancestry and descending lines is possible. For example, for social insect colonies collected in the field, the pedigree is generally unknown. Therefore, only two-generation families are available for the mapping, and information about the linkage phase is missing. Methods for mapping phase unknown have been developed and implemented in a couple of mapping programs (e.g. CRIMAP, Lander & Green, 1987). Since most of these programs are written for small family sizes and diploid organisms like humans, we have developed a simple method to construct linkage maps phase unknown using MAPMAKER version 2.0 (Lander et al., 1987). MAPMAKER has the advantages that it can handle haploid data and that family size is unlimited, features of a mapping population typically for social Hymenoptera. With our new approach it is possible to generate linkage maps for almost every hymenopteran species as long as enough offspring are produced by a single female, which is normally no problem for eusocial Hymenoptera. Additionally, it makes the screening of the grandparents unnecessary and avoids long lasting and difficult breeding experiments, thus saving time and resources.
In this study we generated a linkage map for B. terrestris based on RAPD and microsatellite markers. We used males derived from a queen for which we had no marker information of her parents (=phase unknown). We tested our results derived from our method of mapping phase unknown with a second mapping program (Lander & Green, 1987) that had a mapping phase unknown routine implemented. We could place the sex determining locus on this linkage map and corroborated the existence of a single-locus complementary sex determination system for B. terrestris. Finally, we determined the physical genome size of B. terrestris and could therefore calculate the average amount of recombination frequency per physical unit.
Materials and methods
Origin of bumblebees
All tested individuals (workers, haploid and diploid males) were offspring from one female mated to one of her brothers. Both were derived from the brood of a queen caught in spring 1997 around Zürich, Switzerland. Voucher specimens are stored at the University of Würzburg and ETH Zürich. The mated female hibernated at 5°C in the laboratory for four months. Then she was put into a rearing box (acrylic glass, 12.5 × 7.5 × 5.5 cm) in a climate chamber set at 29°C and 60% relative humidity with a callow worker of another colony to stimulate egg laying. This worker was removed as soon as the young queen had produced five individuals, and the entire incipient colony was transferred into a perlite nest (Pomeroy & Plowright, 1980). The colony eventually produced 57 workers, 409 males (haploid and diploid) and 18 young queens. As expected for diploid male producing colonies, males were produced concurrently with workers. The ploidy status of haploid and diploid males was confirmed with microsatellites (B124–B126, Estoup et al. (1995)) and four codominant RAPD markers (fragment length polymorphism). The mapping population consisted of 116 of the haploid males.
Sample sizes for each experiment
1 Four haploid males of the colony were screened for polymorphism for 1119 RAPD primers and nine microsatellites.
2 One-hundred and nineteen haploid males (i.e. the mapping population) were genotyped using 65 RAPD primers and six microsatellites.
3 Bulk-segregant analysis was performed with six females (workers) and diploid males from the same colony as the haploid mapping population to identify markers linked with the sex determination locus.
4 Forty-eight diploid males and 48 females (workers) from the same colony as the mapping population were tested with markers identified as cosegregating with the sex determination locus.
DNA extraction and RAPD-PCR reactions
DNA from half the thorax of individual males and females was isolated with a standard CTAB-Phenol extraction method (Hunt & Page, 1995). The RAPD-PCR reactions (Williams et al., 1990) were carried out in 12.5 μL reaction volumes using 5 ng of genomic DNA, 0.6 μM primer, 100 μM each of dATP, dCTP, dGTP and dTTP (Pharmacia), 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2 mM MgCl2 and 0.75 U Taq. The 10 nucleotide RAPD primers were obtained from Operon Technologies (Alameda, CA) and the University of British Columbia Biotechnology Centre (Vancouver, Canada). Amplification was performed with the following parameters: five cycles of 94°C/1 min, 35°C/1 min and 72°C/2 min, and another 32 cycles at 94°C/10 s, 35°C/30 s and 72°C/30 s.
Gel electrophoresis and scoring
The amplification products were resolved in 20 × 25 cm horizontal gels using 1% Synergel (Diversified Biotech, Newton Center, MA) and 0.6% Agarose in a 0.5 × TBE buffer. Gels were run for 500–600 Vh, stained in ethidium bromide for 25 min, destained in distilled water for another 40 min, and recorded on an UV transilluminator with Polaroid 667 films. After the map and the linkage groups were established, all markers were ordered according to their position in the linkage groups, and all gels were scored a second time. This allowed us to control for unlikely double crossovers due to scoring errors.
Microsatellite PCR and scoring
The mother (queen) of the mapping population was heterozygous for eight of nine microsatellite loci tested. Six of the heterozygous loci (B10, B11, B116, B118, B124, and B126) were used for further linkage analysis. PCR detection of the microsatellites was done according to Schmid-Hempel & Schmid-Hempel (2000). The reaction mixtures each contained 1–10 ng of total DNA, and multiplex PCRs were run for the loci B10–B11, B116–B118, and B124–B126 (Estoup et al., 1995).
Phase unknown linkage analyses
For mapping it is normally necessary to assign a linkage phase to every marker, i.e. the alleles coming from the grandmother are coded differently from the alleles coming from the grandfather. As the grandparents of the mother (queen of our colony) of our mapping population were not available to determine the linkage phase, we designed the following simple procedure to map phase unknown in MAPMAKER (Lander et al., 1987), that has no phase unknown mapping procedure implemented.
1 First we assigned a phase arbitrarily to every allele of our markers (our convention was to assign 1 to presence alleles or to the longer allele of a fragment length polymorphism, and 0 to absence or to the shorter alleles, respectively).
2 Then for each marker, an artificial second marker was introduced into the data set having the same genotype but complementary phase. Since we had haploid individuals as mapping populations we now had a data set with twice as many markers and each possible linkage phase.
3 MAPMAKER (Lander et al., 1987; version 2.0 for Macintosh; data type was coded as ‘haploid’) was used to do a complete two-point analysis of this doubled dataset. This procedure allowed us to determine the phase and to calculate simultaneously the recombination frequencies between linked markers. Once we knew the phase of the markers we discarded all markers with the wrong phase for the next step, the multipoint analysis. Note, only the high progeny number allowed us to determine the phase of linked markers in our approach.
4 For the following multipoint analysis the phase was known and we could proceed with a normal linkage analysis.
The mapping procedure with MAPMAKER followed a standard protocol described below.
1 A two-point linkage analysis of the doubled dataset (194 markers, see Results) was performed with the ‘GROUP’ command (setting: LOD=3.0; theta=0.4) to find a preliminary set of linkage groups. Note, that this was the doubled data set and that every linkage group had therefore to be present twice. All of the linkage groups we found followed this prediction, i.e. we found 36 identical linkage groups. Using this procedure we determined the linkage phases of the markers. Once phase information was established, multipoint analysis could be carried out with a phase known data-set.
2 Multi-point analysis within all putative linkage groups generated in step 1 were done with the ‘FIRSTORDER’ command (LOD=3.0, theta=0.4). This analysis gave the most likely order of the markers in each linkage group.
3 Using the ‘RIPPLE’ command, the order found in step 2 was tested within each linkage group for all possible three-point orders of consecutive markers. The most likely order for every marker is shown. Markers that were linked at 2 cM or less could not be ordered at a LOD 2 threshold because with the given size of the mapping population there were too few informative meioses.
All map distances (cM) were calculated from recombination fractions (%) according to Kosambi’s mapping function (Kosambi, 1944) because Kosambi’s function resulted in less map expansion than Haldane’s function when the ‘DROP MARKER’ command was used.
Test for the phase unknown mapping procedure – I
We used CRIMAP – a program in which a phase unknown mapping procedure is implemented – to see whether we would get the same result as with our method. To generate a phase unknown data set for CRIMAP we introduced a third non-informative allele for each marker. This simulated a diploid mapping population with an uninformative father, which is necessary to run CRIMAP. Then we did a two-point analysis to generate linkage groups. To overcome the problem that CRIMAP could not be applied to all markers at once, smaller data subsets comprising a smaller number of markers each were formed to be analysed by CRIMAP. The subsets were created in such a way that each pair of markers was included in at least one dataset making sure that linkage between these markers could be detected.
Test for the phase unknown mapping procedure – II
In order to test our phase unknown mapping procedure empirically with a known linkage map we performed two tests with the Apis mellifera map data set which has 365 mapped markers (Hunt & Page, 1995). In the first test we took 100 linked markers (randomly chosen), doubled the data, and reversed phase for the doubled data. Then we ran MAPMAKER (Lander et al., 1987) to see whether we obtained exactly the same linkage groups as in the original map. This test was performed twice.
In the second test we took the markers of the three biggest linkage groups to check whether we would get the same ordering of markers within the linkage groups. Although this was not a rigorous statistical test it demonstrated the power of our mapping phase unknown approach.
Comparing linkage maps and relative map sizes between species
Most of the published maps for non-model organisms are unsaturated (average marker density >10 cM). Therefore, estimates of real map sizes for these species are lacking. However, relative map sizes may be obtained and compared by controlling for marker numbers as a covariate; we can construct maps with equal numbers of markers and compare their sizes. A larger genome would yield a larger map size with the same number of markers. For example, to compare the map sizes of A. mellifera (Hunt & Page, 1995), Nasonia spp. (Gadau et al., 1999), and B. terrestris (this study) the linked markers of all data sets (A. mellifera, 365 markers; Nasonia, 91 markers; and B. terrestris, 79 markers) were randomized and linkage of 20, 40, 60, and 80 (79 in the case of B. terrestris) markers were calculated for each species using MAPMAKER with the default settings. For all species, we then calculated the total map size as the size of each linkage group plus 40 cM to account for all unlinked markers. Our justification for adding 40 cM is that all markers must be linked somewhere, but were left unlinked, during the mapping procedure, because they had no companion markers within 40 cM of them (given by the setting of theta=0.40). Our expectation is that genomes with overall higher rates of recombination will be relatively much larger. This is because fewer markers are likely to be linked together into a single linkage group if the linkage map of a species is large. This effect is expected to be particularly evident when few markers are used. However, as marker numbers increase, linkage groups should coalesce, resulting in a slower increase in map size as the map approaches saturation.
Physical genome size of B. terrestris
To determine the physical genome size of B. terrestris we conducted standard flow cytometry of thoracic muscle cells (for detailed methodology see Otto, 1994). As size standard we used thoracic muscle cells of A. mellifera since the physical size of the haploid genome of A. mellifera is known to be 178 Mb (Jordan & Brosemer, 1974; Hunt & Page, 1995).
Mapping of the sex locus
To find markers segregating for the sex determination locus we combined the DNA of six diploid males and six workers (derived from the same queen as the haploid males of the mapping population) into separate samples (= bulk). We screened for any marker present in the diploid males but absent in the workers and vice versa (‘bulk segregation analysis’, e.g. De Tomaso et al., 1998). Markers segregating between diploid males and workers were independently tested with six different individual diploid males and workers, respectively. Markers which again segregated consistently between diploid males and workers were tested on an additional 48 diploid males and 48 workers to confirm linkage with the sex locus and to derive an estimate of the distance between these markers and the sex locus. Every marker which shows variability between the diploid males and workers must also be variable in the haploid males of the same colony, because the queen must be heterozygous for this marker. Therefore, having identified markers linked to the sex locus, they immediately could be integrated into the linkage map derived from the 116 males of the mapping population.
Results
Linkage map
A total of 1119 RAPD primers (Operon primer sets A–Z and UBC primers 1–599.) were prescreened on four haploid males of the mapping population. The best 65 RAPD primers generated 91 segregating nuclear markers; that is an average of 1.4 markers per RAPD primer. Additionally, six microsatellite markers segregated in the mapping population. Of the RAPD markers 71 (78%) showed presence/absence polymorphism whereas 20 markers (22%) displayed fragment length polymorphisms. The recovery rate of all alleles was normally distributed (mean ± SD: 0.50 ± 0.07; Fig. 1)

Plot of the recovery rates (number of individuals with allele 1/number of all individuals tested) for all markers. The recovery rate is normally distributed (mean ± SD=0.50 ± 0.07).
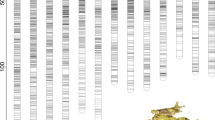
MAPMAKER (LOD 3.0; theta 0.40; Lander et al., 1987) mapped 79 of 97 markers (82%) into 21 linkage groups (Fig. 2) The map spanned 953.1 cM with an average marker spacing of 12.1 cM. Since B. terrestris has 18 chromosomes (Hoshiba et al., 1995) three of the 21 linkage groups must be linked with other linkage groups. Thus, the total genome size was estimated to be 1073 cM, assuming that three of the linkage groups have to be linked and that gaps represent at least 40 cM due to our setting of MAPMAKER (theta=0.40).

Linkage map of B. terrestris based on RAPD markers. Markers are named by their primer designation. The primer designation is a letter and a number for primers of Operon Technologies, or a number for primers from the University of British Columbia. The primer name is followed by a dash and the approximate size of the amplified fragment in kilobases. Markers showing fragment-length polymorphisms are indicated with an “f” following the marker name. The position of the sex locus was determined by comparing the distance between the two markers 389-0.36 (7.5 cM) and 237-0.30 (16.5 cM) and the phenotypic sex in diploid males and workers, and the distance of these markers in haploid males (24.1 cM). The sex locus must be located between these markers because 16.5 cM and 7.5 cM (the distance between the sex determination locus and the markers) sum to 24.0 cM.
Test for the phase unknown mapping procedure – I
The two-point linkage analyses with CRIMAP produced the exact same linkage groups as we did with our approach with MAPMAKER (doubling the dataset and flip-phase). However, there were three markers, V17-0.87 (LG I), 570-0.55 (LG II) and 465-0.54 (LG IV) which were linked, with LOD scores smaller than 3.0 (2.75, 2.84, and 2.93, respectively) but had higher LOD scores when we used our approach with MAPMAKER (3.05, 3.14, and 3.23, respectively). In general all LOD scores in CRIMAP were 0.3 units smaller than the LOD scores in our MAPMAKER approach (results not shown). This was due to the following differences in calculating the LOD scores in CRIMAP and MAPMAKER.
MAPMAKER calculated the LOD scores simply by using the likelihood ratio:

where θ=recombination fraction; n=number of individuals genotyped; n(rec)=number of putative recombinants; n(non-rec)=number of putative non-recombinants), because although we generated two additional markers (by doubling and phase-switching) the two-point LOD score calculation only uses the information of two loci at a time.
In contrast, CRIMAP uses the more appropriate formula for the likelihood ratio:

This formula is more appropriate because it combines the probabilities for both possible phases in the calculation of the L(theta).
If n is large the likelihood ratio calculated by MAPMAKER is twice that computed by CRIMAP, which leads to a mapmaker LOD score that is inflated by log10 2=0.3.
To account for the overestimation of the LOD scores in our approach we could simply increase our LOD threshold by 0.3 units. However, since the size of the linkage map of B. terrestris is only around 1000 cM, an appropriate LOD threshold for the linkage of two markers in B. terrestris would be 2.5 (see p. 68 in Ott, 1999 for a discussion of LOD thresholds). Therefore, using a LOD score threshold of 3.0 with our approach already compensates for this deviation.
Once we have established the phase of a marker we can simply discard the markers with the “wrong” phase and proceed with a multipoint analysis in the ‘usual way’ (see Materials and methods).
Testing the phase unknown mapping procedure – II
Using our approach with the 395 RAPD marker set of Apis mellifera (Hunt & Page, 1995) we generated the same linkage groups and the same order of markers within the linkage groups as in the original map of Hunt & Page (1995) where phase was known.
Mapping of the sex determination locus
We screened 499 primers (UBC primers 100–599) in the bulk segregation analysis of two DNA template mixtures of six diploid males and six workers each. Two markers (237-0.30 and 389-0.36) differed between bulks and also segregated during the screening of six different individual workers and six diploid males. A consecutive run on 48 diploid males and 48 workers each revealed that both markers were linked to the sex locus (237-0.30=16.5 cM and 389-0.36=7.5 cM). Since the markers also segregated in the haploid males of the mapping population, the sex locus could be incorporated into the linkage map. Only one sex determination locus was found and it mapped into a single linkage group IX (Fig. 2), together with five other markers (three additional Operon markers which were previously scored for the haploid males only, see Fig. 2). Only one of these three Operon markers (Z7-4.0) could also be mapped in the diploid workers and males. The other two Operon markers could not be scored in diploid individuals because the mate of the queen had the present allele. Note, that RAPD markers are generally dominant markers so that in diploid individuals heterozygotes of the allelic type present/absent cannot be distinguished from homozygotes of the type present/present. The most likely position of the sex locus is given in Fig. 2. The distances between the flanking markers (389-0.36 and 237-0.30) of the sex locus in linkage group IX, derived from the diploid individuals and the haploid mapping population, were almost identical (24.1 cM vs. 24 cM, results not shown). However, the distances between the marker 237-0.30 and Z7-0.49 were quite different in the haploid and diploid map (10.8 vs. 1.1 cM).
Physical genome size of B. terrestris
Thoracic muscle cells of B. terrestris contained 1.54 times more DNA than the cells of A. mellifera (N=50 000 cells, coefficients of variance (%) for A. mellifera and B. terrestris were 2.53 and 2.52, respectively). The physical size of the haploid genome of B. terrestris was thus calculated to be 274 Mb (=1.54 × 178 Mb).
Discussion
Genome map
Bombus terrestris is the second social insect species for which a linkage map is now available. The map contains 79 markers and spans over 953.1 cM. The minimal recombinational size of the B. terrestris genome was estimated to be 1073 cM. The linkage map of B. terrestris is thus significantly smaller than the linkage map of the closely related honey bee (Apis mellifera) but it is similar to other hymenopteran species (Fig. 3) On the other hand, the physical genome of B. terrestris was estimated to be 274 Mb and is therefore 1.54 times larger than that of A. mellifera (178 Mb). Therefore, 1 cM in the linkage map of B. terrestris equals on average 255 kb in physical units. In contrast 1 cM in the honey bee map equals 50 kb (Hunt & Page, 1995). This means that on average A. mellifera has an astonishing five times more recombinational events per kb than B. terrestris (see also Fig. 3). This is especially interesting because A. mellifera belongs to the same family, Apidae, as B. terrestris but has one of the highest recombination frequencies ever reported for higher eukaryotes (Hunt & Page, 1995). We now can exclude several structural explanations like haplo-diploidy or small chromosomes for this high recombination frequency in A. mellifera, as originally proposed by Hunt & Page (1995), because both A. mellifera and B. terrestris have a single-locus CSD system and both have small chromosomes (Hoshiba et al., 1995). We can also exclude that the evolution of sociality favours an increase in recombination frequency per se because B. terrestris is eusocial as well. Rather, it appears more likely that ‘external’, ecological reasons have driven the evolution of high recombination frequencies in A. mellifera. For example, the high recombination frequency of A. mellifera could be associated with the more complex and elaborated system of division of labour in A. mellifera (Page & Robinson, 1991) or with high parasite loads in this species (Otto & Michalakis, 1998; Schmid-Hempel, 1998). Division of labour is thought to profit from an increased recombination because it increases the genotypic and presumably phenotypic diversity among workers within a colony for multigenic traits, assuming that these variable genes are linked together on chromosomes. This could lead to a more stable division of labour. If division of labour is a determinant of the high recombination frequency in A. mellifera, we would expect that closely related species like A. dorsata and very distantly related eusocial hymenopteran species with a very similar type of division of labour like the leaf cutter ants (e.g. species from the genus Acromyrmex) should have equally high recombination frequencies. To evaluate this hypothesis we are currently working on linkage maps for Acromyrmex echinatior. Alternatively, parasites and pathogens can theoretically produce an increase in recombination (Peters & Lively, 1999). An increase of recombination would produce a genetically more diverse worker force and might so hamper the colonization of a whole colony by a single pathogen or parasite genotype.

Comparison of relative map sizes between Apis mellifera, Bombus terrestris, and three parasitic Hymenoptera (Bracon hebetor, Antolin et al., 1996; Trichogramma brassicae, Laurent et al., 1998; Nasonia spp., Gadau et al., 1999). Dashed lines are the 95% confidence limits of the regression line. It is very obvious that A. mellifera has a significantly higher recombination frequency than all other hymenopteran species for which linkage maps have been published.
Mapping of the sex locus
The bulk segregation analysis of diploid workers and diploid males was a quick and efficient way to find molecular markers linked with the sex locus in B. terrestris. Since the diploid individuals were produced by the same female (queen) as the haploid mapping population we could directly incorporate the molecular markers linked with the sex locus into the linkage map of B. terrestris.
Our results (Fig. 2) support a single-locus sex determination mechanism in B. terrestris. (Duchateau et al., 1994), and makes a two-locus sex determination system as proposed for B. atratus, a neotropical bumblebee (Garofalo, 1973) unlikely. Alternatively, it is possible that in B. atratus different selection pressures like high inbreeding frequencies might have favoured the evolution of a multilocus CSD system, but to our knowledge nothing is known about inbreeding frequencies in this species.
However, we cannot exclude the possibility of additional QTL (quantitative trait loci) influencing the sex determination in B. terrestris, as shown by Holloway et al. (2000) for a Bracon sp. near hebetor, because we have not done a QTL analysis of the sex determination of the diploid individuals — workers and diploid males — for all markers. However, both, breeding studies and the molecular data now are consistent with a single-locus CSD mechanism for B. terrestris.
The distance between the sex locus and the closest marker of our linkage map is 7.5 cM, i.e. approximately 1.9 Mb in physical units. This is prohibitively large for identifying the gene directly by genome walking, but it will be the starting point in future studies to search for markers associated more closely with the sex locus. Clearly, a knowledge of the genomic map, including the sex locus, in B. terrestris has a variety of exciting applications, from the possibility of characterizing sex genes to studies of selection and maintenance of allelic diversity at the population level.
A linkage map of B. terrestris containing 79 markers and which is produced by a newly designed phase unknown mapping procedure is sufficient to map a single-locus trait — here the sex determination locus — by using a bulk segregation approach. This result is promising for future projects in B. terrestris, including mapping of quantitative traits like disease resistance or behaviour. In general this new mapping approach combined with highly variable anonymous molecular markers (AFLP, RAPD, etc.) opens a new avenue towards a fast and efficient mapping of a non-model organism which can not be bred in the laboratory, as is the case for most social Hymenoptera.
References
Antolin, M. F., Bosio, C. F., Cotton, J., Sweeney, W. et al., (1996). Intensive linkage mapping in a wasp (Bracon hebetor) and a mosquito (Aedes aegypti) with Single-Strand Conformation Polymorphism analysis of Randomly Amplified Polymorphic DNA markers. Genetics, 142: 1727–1738.
Beye, M., Moritz, R. F. A., Crozier, R. H. and Crozier, Y. C. (1996). Mapping the sex locus of the honey bee (Apis mellifera). Naturwissenschaften, 83: 424–426.
Bull, J. J. (1983). Evolution of Sex Determining Mechanisms. Cummings, Menlo Park, CA.
Butcher, R. D. J., Whitfield, W. G. F. and Hubbard, S. F. (2000). Single locus complementary sex determination in Diademan chrysotictes (Gmelin) (Hymenoptera: Ichneumonidae). J Hered, 91: 104–111.
Cook, J. M. (1993). Sex determination in the Hymenoptera: a review of models and evidence. Heredity, 71: 421–435.
Crozier, R. H. (1977). Evolutionary genetics of the Hymenoptera. Ann Rev Entomol, 22: 263–288.
de Tomaso, A. W., Saito, Y., Ishizuka, K. J., Palmeri, K. J. et al., (1998). Mapping the genome of a model protochordate. I. A low resolution genetic map encompassing the fusion/histocompatibility (Fu/HC) locus of Botryllus schlosseri. Genetics, 149: 277–287.
Duchateau, M. J., Hoshiba, H. and Velthuis, H. H. W. (1994). Diploid males in the bumblebee Bombus terrestris. Entomol Exp Appl, 71: 263–269.
Dzierzon, J. (1845). Gutachten über die von Herrn Direktor Stöhr im ersten und zweiten Kapitel des General-Gutachtens aufgestellten Fragen. Eichstädter Bienenzeitung, 1, 109–113, 119–121.
Estoup, A., Scholl, A., Pouvreau, A. and Solignac, M. (1995). Monandry and polyandry in bumblebees (Hymenoptera; Bombinae) as evidenced by highly variable microsatellites. Mol Ecol, 4: 89–93.
Gadau, J., Page, R. E., Jr and Werren, J. H. (1999). Mapping of hybrid incompatibility loci in Nasonia. Genetics, 153: 1731–1741.
Garofalo, C. A. (1973). Occurrence of diploid drones in a neotropical bumblebee. Experientia, 29: 726–727.
Holloway, A. K., Strand, M. R., Black, W. C., IV and Antolin, M. F. (2000). Linkage Analysis of sex determination in Bracon sp. near hebetor (Hymenoptera: Braconidae). Genetics, 154: 205–212.
Hoshiba, H., Duchateau, M. J. and Velthuis, H. H. W. (1995). Diploid males in the bumblebee Bombus terrestris (Hymenoptera): karyotype analyses of diploid females, diploid males and haploid males. Jap J Ent, 63: 203–207.
Hunt, G. J. and Page, R. E. (1994). Linkage analysis of sex determination in the honey bee (Apis mellifera). Mol Gen Genet, 244: 512–518.
Hunt, G. J. and Page, R. E., JR . (1995). Linkage map of the honey bee, Apis mellifera, based on RAPD markers. Genetics, 139: 1371–1382.
Jordan, J. R. and Brosemer, R. W. (1974). Characterization of DNA from three different bee species. J Insect Physiol, 20: 2513–2520.
Kosambi, D. (1944). The estimation of map distances from recombination values. Ann Eugen, 12: 172–175.
Lander, E. S. and Green, J. (1987). Construction of multilocus genetic linkage maps in human. Proc Natl Acad Sci USA, 84: 2363–2367.
Lander, E. S., Green, P., Abrahamson, J. and Barlow, A. et al. (1987). MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics, 1: 174–181.
Laurent, V., Wajnberg, E., Mangin, B. and Schiex, T. et al. (1998). A composite genetic map of the parasitoid wasp Trichogramma brassicae based on RAPD markers. Genetics, 150: 275–282.
Ott, J. (1999). Analysis of Human Genetic Linkage, 3rd edn. The Johns Hopkins University Press, Baltimore.
Otto, F. J. (1994). High resolution analysis of nuclear DNA employing the fluorochrome DAPI. In: Darzynkiewicz, Z., Robinson, J. P. and Crissman, A. (eds) Methods in Cell Biology, pp. 211–217. Academic Press, San Diego.
Otto, S. P. and Michalakis, Y. (1998). The evolution of recombination in changing environments. Trends Ecol Evol, 13: 145–151.
Page, R. E. and Robinson, G. E., JR . (1991). The genetics of division of labor in honey bee colonies. Adv Insect Physiol, 23: 117–169.
Peters, A. D. and Lively, C. M. (1999). The Red Queen and fluctuating epistasis: a population genetic analysis of antagonistic coevolution. Am Nat, 154: 393–405.
Pomeroy, N. and Plowright, R. C. (1980). Maintenance of bumblebee colonies in observation hives (Hymenoptera: apidae). Can Entomol, 112: 321–326.
Schmid-Hempel, P. (1998). Parasites in Social Insects. Princeton University Press, Princeton, NJ.
Schmid-Hempel, R. and Schmid-Hempel, P. (2000). Female mating frequencies in social insects: Bombus spp. from Central Europe. Insectes Soc, 47: 36–41.
Whiting, P. W. (1943). Multiple alleles in complementary sex determination of Habrobracon. Genetics, 28: 365–382.
Williams, J. G. K., Kubelik, A. R., Livak, K. K., Rafalski, J. A. et al., (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucl Acids Res, 18: 6531–6335.
Woyke, J. (1963). What happens to diploid drones in a honey bee colony? J Apic Res, 2: 73–76.
Acknowledgements
This work was supported by a Feodor-Lynen Forschungsstipendium and the Deutsche Forschungsgemeinschaft (GA 661) to JG, a grant by the Swiss NSF to PSH (nr. 3100–049040.95) and a grant to REP (PHS-MH5311). We thank C. Steinlein and M. Schmid for their help with the flow cytometry. We thank two anonymous reviewers for comments on this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gadau, J., Gerloff, C., Krüger, N. et al. A linkage analysis of sex determination in Bombus terrestris (L.) (Hymenoptera: Apidae). Heredity 87, 234–242 (2001). https://doi.org/10.1046/j.1365-2540.2001.00919.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.2001.00919.x
Keywords
This article is cited by
-
QTL mapping identifies a major locus for resistance in wheat to Sunn pest (Eurygaster integriceps) feeding at the vegetative growth stage
Theoretical and Applied Genetics (2017)
-
High-density sex-specific linkage maps of a European tree frog (Hyla arborea) identify the sex chromosome without information on offspring sex
Heredity (2016)
-
Establishment risk of the commercially imported bumblebee Bombus terrestris dalmatinus—can they survive UK winters?
Apidologie (2016)
-
Expression profile of the sex determination gene doublesex in a gynandromorph of bumblebee, Bombus ignitus
The Science of Nature (2016)
-
Population-level consequences of complementary sex determination in a solitary parasitoid
BMC Evolutionary Biology (2015)
