
Abstract
This paper addresses a decades-old taxonomic controversy surrounding a species in the grasshopper subfamily Melanoplinae. Melanoploid grasshoppers fall into two tribes, the Nearctic-restricted Melanoplini and the Holarctically distributed Podismini. The current view regarding one member, Bohemanella frigida, is that it belongs to the latter tribe and that North American populations were established by dispersal from Eurasia via the Bering Land Bridge. Over the past 50 years, this opinion has changed a few times; this species was once regarded as part of the tribe Melanoplini and, as such, deemed to be the only Holarctically distributed Orthopteran insect with New World antecedents. A molecular phylogenetic study of this species was thus performed to verify its phylogenetic position and to establish a probable direction of dispersal. Portions of three mitochondrial genes (cyt b, COII, and ND2) were sequenced and phylogenetically analysed using weighted and unweighted parsimony, neighbour-joining, and maximum likelihood methods. Support for the inclusion of B. frigida within the tribe Melanoplini and the use of its original name, Melanoplus frigidus, was strong using all methods. Placement in the tribe Melanoplini leads to an acceptance of an earlier hypothesis regarding direction of dispersal across the Bering Land Bridge, making this grasshopper a unique case in orthopteran insects in this respect.
Similar content being viewed by others


Introduction
Among many issues surrounding the systematics of a large subfamily of acridid grasshoppers, the Melanoplinae (Vickery, 1987), are the taxonomic position and biogeographic origin of one member, Bohemanella frigida. Were it not once regarded as the only known example of a Holarctically distributed orthopteran insect, but with Nearctic ancestors (Vickery & Kevan, 1983), the species would barely pass scrutiny, owing to a rather indistinctive morphology.
Following Vickery (1987), melanoploid grasshoppers fall into two tribes, Melanoplini and Podismini. [There are other classification schemes (Rehn & Randell, 1963; Vickery, 1997), but we shall follow this one]. The Melanoplini are restricted to the Nearctic region, whereas the Podismini occur throughout the Palaearctic and in several Nearctic locales (Vickery, 1987). B. frigida is currently distributed at high altitudes or northern latitudes within Eurasia, while in North America it occurs in Alaska, the Northwest Territories, and the Yukon (Vickery & Kevan, 1983). Originally named Melanoplus frigidus, this species was included in the tribe Melanoplini along with all other members of this genus. B. frigida acquired its present name on the basis of a supposedly peculiar structure of the mesosternum (Ramme, 1951). Mishchenko (1952), however, regarded the structure as quite variable and insufficiently distinct to warrant removing the taxon from Melanoplus. Mishchenko went on to state that there were no other distinguishing morphological features which would justify changing this insect’s taxonomic affiliation. Nevertheless, Vickery (1987), citing unpublished work by N. Jago, made the decision to regard the grasshopper as B. frigida and as a member of the Podismini. Skareas & Hsiung (1999), in a recent analysis comparing several male genital traits among B. frigida and a number of podismine and melanopline taxa, claimed additional support for this view, although they admitted that such traits were highly variable. In any case, the current interpretation with respect to biogeographical origin is that Nearctic populations of B. frigida were established by dispersal from the Palaearctic via the Bering Land Bridge (Vickery, 1987).
To address this B. frigida/M. frigidus conundrum, and perhaps help infer a probable direction of intercontinental dispersal, a molecular phylogenetic analysis involving North American and Eurasian melanoplines was undertaken. Mitochondrial DNA (mtDNA) has features (reviewed by Harrison, 1989) making it suitable for use in this study; portions of the molecule were sequenced for this purpose.
Materials and methods
Six ethanol-preserved or museum specimens of B. frigida, taken from several locations in this insect’s range, were analysed (Table 1). To assess B. frigida’s connection with the two tribes of Melanoplinae (Melanoplini and Podismini), six members of the genus Melanoplus (sanguinipes, bivittatus, infantilis, packardii, marginatus, and microtatus) were analysed as well as three podismine species (Podisma pedestris, Zubovskya koeppeni, and Miramella alpina). The species Melanoplus marginatus and M. microtatus are members of a supposedly primitive Melanoplus species group Marginatus (Rentz, 1978; Chapco et al., 1999). A list of specimens, source of organisms, tissue analysed, and location collected is provided in Table 1. Outgroup taxa were Schistocerca gregaria, a member of the subfamily Cyrtacanthacridinae, and Locusta migratoria, a member of the subfamily Oedipodinae, and supposedly more distantly related (Chapco et al., 1997). An association between Schistocerca and Melanoplus is presumed since both were at one time assigned to the same subfamily, Cyrtacanthacridinae (Vickery & Kevan, 1983). A recent report based on mtDNA sequences (Chapco et al., 1999) confirmed the suitability of Schistocerca as an outgroup for melanopline studies.
Depending on the method of storage and age of the specimen, total DNA was extracted using a standard phenol procedure (Chapco et al., 1992), a ‘boiling method’ (Hoy, 1994), or a method using DTAB and CTAB (Phillips & Simon, 1995). DNA extraction from museum specimens was most successful using the method of Phillips & Simon (1995).
Regions of three mitochondrial genes, cytochrome b (cyt b), cytochrome oxidase subunit II (COII), and NADH dehydrogenase subunit II (ND2) were analysed. Primers used for amplification and sequencing are given in Table 2. Primers marked with an asterisk (*) were either designed using published L. migratoria sequences (Accession Number X80245) (Flook et al., 1995) or, for COII, part of a published list (Simon et al., 1994). These primers were used to amplify longer fragments of the various genes from freshly collected insects. For some older museum specimens, however, amplification of these longer fragments was unsuccessful and primers internal to those marked with an asterisk were used in order to amplify overlapping shorter portions of the various genes (Table 2). For cyt b and COII, these internal primers were designed using the respective Melanoplus sanguinipes sequences (Chapco et al., 1999) with the exception of the mtD16 primer, known also by the title C2-J-3400 (Simon et al., 1994). Similarly, internal primers for the ND2 gene were designed using a consensus sequence obtained from various Melanoplus species.
PCR amplification was performed in a 50 μL volume containing 2.6 units of Expand High Fidelity enzyme (Boehringer Mannheim), 1× Expand PCR Buffer 2, 50 μM each dNTP (Boehringer Mannheim), 500 nM each primer and 2 μL of purified DNA (when using the extraction method of Phillips & Simon, 1995) or 37.5 ng genomic DNA (using other extraction methods). Amplifications were performed using a GeneE Thermal Cycler (Techne). Amplification conditions were as follows: an initial denaturation of 94°C for 2 min followed by 10 cycles of 94°C for 10 s, 45°C for 30 s, and 68°C for either 30 s (shorter fragments from museum specimens), 45 s (cyt b and COII), or 50 s (ND2). For the final 25 cycles (30 cycles for museum specimens), cycling was identical to the first 10 cycles except that the annealing temperature was raised to 50°C and an additional 10 s was added to the primer extension step each cycle. A final extension step of 68°C for 3 min followed. PCR products were either purified directly using the Wizard PCR Preps DNA Purification System (Promega) or, after being resolved in an agarose gel and the appropriate band excised with a scalpel, purified using the Wizard PCR Preps DNA Purification System (Promega) or Qiagen Gel Extraction Kit (QIAGEN), depending on availability.
Following manufacturer’s recommendations, DNA sequencing was performed either manually using the T7Sequencing Kit (Pharmacia Biotech) or the ABI Prism 377 DNA automated sequencing system at the Plant Biotechnology Institute, Saskatoon, SK.
Sequences were easily aligned by visual inspection and imported into MACCLADE (Maddison & Maddison, 1992). Phylogenetic relationships were inferred using maximum parsimony (MP) and neighbour-joining (NJ) methods, available in the software package PAUP, version 4.0b4a (Swofford, 1999). MP searches were conducted employing simple addition sequences in conjunction with the branch and bound algorithm. In order to decrease the possible effects of homoplasy in the data set, successive rounds of weighting according to Farris’s (1969) scheme (wMP) were performed using, as weights, rescaled consistency indices. For distance-based analyses, pairwise sequence differences were compared using the Kimura 2-parameter (K2) transformation. Levels of support for trees derived from MP, wMP and NJ analyses were estimated by performing 1000 bootstrap replicates. Bootstrap values for both MP and wMP analyses were obtained using the branch and bound algorithm.
A partition homogeneity test (Farris et al., 1995), available in PAUP (Swofford, 1999), was undertaken to assess whether different gene regions yielded significantly different topologies. This test was performed using 1000 random partitions of combined gene region data sets.
The likelihoods of trees representing competing biogeographic hypotheses with respect to the tribe affiliation of B. frigida were compared using the Kishino–Hasegawa test (Kishino & Hasegawa, 1989), also available in PAUP (Swofford, 1999).
Results and discussion
DNA sequence data from these insects comprised approximately 1106 bases, consisting of 258 bases from the cyt b gene, 381 bases from the COII gene, and 467 bases from the ND2 gene. Sequences have been deposited in GenBank under Accession Numbers AF145491, AF145499, AF145500, AF145508, AF145509, AF145520, AF145521, AF145523, AF145524, AF145560, AF145561 and AF227276–AF227311, inclusive. DNA bracketed by the primers ND2A and ND2C failed to amplify in B. frigida specimens from Russia, Alaska, and the Northwest Territories as well as Zubovskya and Melanoplus microtatus. This failure resulted in the loss of approximately 110 bases of the ND2 data set for these specimens; these positions were treated as unknown in parsimony analyses and ignored for pairwise comparisons in distance-based analyses. Across the three genes, 204 sites were phylogenetically informative. High A + T contents were noted for the mtDNA of these grasshoppers, in agreement with values for other melanoplines (Chapco et al., 1999) and insects in general (Simon et al., 1994). Average A + T contents were 69.8%, 68.4%, and 73.8% for cyt b, COII, and ND2 genes, respectively.
Tests of homogeneity between the various gene sequences indicated significant heterogeneity in the unweighted data set (P=0.008), whereas this was not the case (P=0.196) when sites were weighted according to Farris’s (1969) successive approximations weighting scheme (see below). The data were therefore analysed simultaneously. In general, greater levels of resolution were attained; relationships not evident in single-gene trees were observed, a result noted by other investigators (Baker & DeSalle, 1997; Crespi et al., 1998).
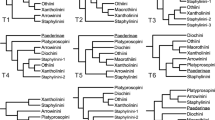
MP analysis of the unweighted data set yielded four most parsimonious trees which differed at lower taxonomic levels (tree length=508, consistency index= 54.1%). Weighted MP analysis required three iterative rounds to obtain stable weights, returning one tree with the same topology as one of the aforementioned four (tree length=155.086, consistency index=73.3%). Figure 1 is a consensus of MP, wMP and NJ-derived trees. The three differ in minor ways, notably in the positioning of various species within Melanoplus and the position of individuals in the B. frigida clade; however, the incongruity does not detract from the major focus of this paper. Differences between the MP, wMP and NJ analyses are depicted as unresolved branches in the consensus tree. Included are bootstrap values for all three procedures.

Consensus of MP, wMP and NJ-K2 analyses. Bootstrap levels (1000 replicates) for unweighted parsimony (above branches, left of semicolon), weighted parsimony (above branches, right of semicolon), and NJ-K2 (below branches) are given. The asterisk refers to bootstrap levels <50%. Note: Branch lengths are not to scale.
The most striking outcome of this analysis is that B. frigida is strongly placed (bootstrap levels of 97% for MP and 100% for both wMP and NJ-K2) among members of the tribe Melanoplini (Fig. 1) rather than with members of the Podismini, a result contrary to the current viewpoint (Vickery, 1987; Skareas & Hsiung, 1999). Furthermore, a comparison of the likelihoods of the wMP tree and a tree where the B. frigida clade was repositioned to reflect a taxonomic affiliation with the Podismini provides high statistical support for the association with the Melanoplini (Δln L=117.693 ± 25.383; P < 0.0001). This positioning with the Podismini also added 34 steps to the length of the most parsimonious tree. A reversion to the original name, M. frigidus, should therefore be considered. It is to be noted that a recent analysis of 610 bases of the mitochondrial cytochrome oxidase subunit I gene sequenced in a subset of the present taxa (data not shown) also supports a close affiliation within the Melanoplini.
The value of a phylogenetic approach to biogeographic issues is its potential for deducing, for example, direction of dispersal (Zink et al., 1995). Given that all analysed populations of B. frigida are topologically internal within the Melanoplus clade and that the genus is exclusively North American, the weight of evidence supports the conclusion that this species migrated from the New World to Eurasia via the Bering Land Bridge, a view previously expressed (Vickery & Kevan, 1983). The precise sequence of dispersal events is, however, difficult to ascertain owing to poor resolution within the B. frigida clade (Fig. 1). Also, log-likelihood values associated with trees having different permutations of B. frigida populations are not significantly different from one another, indicating that the data can not resolve relationships at this level. Additional sequences, perhaps of nuclear genes, are clearly required.
These analyses may be compromised if any of the sequences have been incorporated into the nuclear genome, a phenomenon recently described for a number of species of Acrididae (Bensasson et al., 2000) and a wide range of vertebrate and invertebrate taxa (Zhang & Hewitt, 1996). Zhang & Hewitt (1996) proposed a list of five practical considerations for ensuring that the studied sequences are mitochondrial. Our sequences appear to satisfy four of these. For example, all PCR amplifications yielded single rather than multiple bands, the latter a predicted outcome if both mitochondrial and nuclear genomes were to contain related sequences differing slightly in size. It is difficult to address the fifth consideration, pertaining to ‘unusual or contradictory’ tree topologies, because the purpose of this study was to determine the phylogenetic position of Bohemanella relative to other melanoplines and there were no a priori predictions concerning this. However, it should be noted that, when Bohemanella was excluded, there was a clean separation of the two melanopline tribes (Fig. 1), in accordance with the conventional dogma (Vickery, 1987). Finally, if some of the sequences had been nuclear and undergone nucleotide insertions or deletions (Zhang & Hewitt, 1996), sequence alignment with respect to L. migratoria, whose entire mtDNA sequence is known (Flook et al., 1995), would have been problematic.
References
Baker, R. H. and Desalle, R. (1997). Multiple sources of molecular characteristics and the phylogeny of Hawaiian drosophilids. Syst Biol, 46: 654–673.
Bensasson, D., Zhang, D. X. and Hewitt, G. M. (2000). Frequent assimilation of mitochondrial DNA by grasshopper nuclear genomes. Mol Biol Evol, 17: 406–415.
Chapco, W., Kelln, R. A. and McFadyen, D. A. (1992). Intraspecific mitochondrial DNA variation in the migratory grasshopper, Melanoplus sanguinipes. Heredity, 69: 547–557.
Chapco, W., Martel, R. K. B. and Kuperus, W. R. (1997). Molecular phylogeny of North American band-winged grasshoppers (Orthoptera: Acrididae). Ann Entomol Soc Am, 90: 555–562.
Chapco, W., Kuperus, W. R. and Litzenberger, G. (1999). Molecular phylogeny of melanopline grasshoppers (Orthoptera: Acrididae): The genus Melanoplus. Ann Entomol Soc Am, 92: 617–623.
Crespi, B. J., Carmean, D. A., Mound, L. A. and Worobey, M. (1998). Phylogenetics of social behavior in Australian gall-forming thrips: Evidence from mitochondrial DNA sequence, adult morphology and behavior, and gall morphology. Mol Phylogenet Evol, 9: 163–180, 10.1006/mpev.1997.0449.
Farris, J. S. (1969). A successive approximations approach to character weighting. Syst Zool, 18: 374–385.
Farris, J. S., Källersjö, M., Kluge, A. G. and Bult, C. (1995). Constructing a significance test for incongruence. Syst Biol, 44: 570–572.
Flook, P. K., Rowell, C. H. F. and Gellissen, G. (1995). The sequence, organization and evolution of the Locusta migratoria mitochondrial DNA genome. J Mol Evol, 41: 928–941.
Harrison, R. G. (1989). Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol Evol, 4: 6–11.
Hoy, M. A. (1994) Insect Molecular Genetics. An Introduction to Principles and Applications. Academic Press, New York, NY.
Kishino, H. and Hasegawa, M. (1989). Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J Mol Evol, 29: 170–179.
Liu, H. and Beckenbach, A. T. (1992). Evolution of the mitochondrial cytochrome oxidase II gene among 10 orders of insects. Mol Phylogenet Evol, 1: 41–52.
Maddison, W. P. and Maddison, D. R. (1992). MACCLADE (version 3.0). analysis of phylogeny and character evolution. Sinauer, Sunderland, MA.
Mishchenko, L. L. (1952) Fauna of the U.S.S.R., vol. IV, no. 2. Leningrad.
Phillips, A. J. and Simon, C. (1995). Simple, efficient, and nondestructive DNA extraction protocol for arthropods. Ann Entomol Soc Am, 88: 281–283.
Ramme, W. (1951). Zur Systematik, Faunistik und Biologie der Orthopteren von sudost-Europa und Vorderasien. Mitt Zool Mus Berl, 27: 1–431.
Rehn, J. A. G. and Randell, R. L. (1963). A preliminary analysis of the lines of the supertribe Melanoplini (Orthoptera; Acrididae; Cyrtacanthacridinae). Proc Natl Acad Sci (Philadelphia), 115: 1–32.
Rentz, D. C. F. (1978). The grasshoppers of the marginatus group of the genus Melanoplus (Orthoptera: Acrididae; Melanoplinae). Acrida, 7: 165–196.
Simon, C., Frati, F., Beckenbach, A. and Crespi, B. (1994). Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am, 87: 651–701.
Skareas, S. D. and Hsiung, C. -C. (1999). The identity of Bohemanella frigida frigida (Boheman) (Orthoptera: Acrididae). Lyman Entomol Museum Res Lab Note, 26: 1–20.
Swofford, D. L. (1999) PAUP*: phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer, Sunderland, MA.
Vickery, V. R. (1987). The northern Nearctic Orthoptera: their origins and survival. In: Baccetti, B. C. (ed.) Evolutionary Biology of Orthopteroid Insects, pp. 581–591. Ellis Horwood Limited, West Sussex, England.
Vickery, V. R. (1997). Classification of the Orthoptera (sensu stricto) or Caelifera. In: Gangwere, S. K., Muralirangan, M. C. and Muralirangan, M. (eds) The Bionomics of Grasshoppers, Katydids, and Their Kin, pp. 5–40. Cab International, New York.
Vickery, V. R. and Kevan, D. K. (1983) A Monograph of the Orthopteroid Insects of Canada and Adjacent Regions. Lyman Entomological Museum and Research Laboratory Memoir, 13, 1–1462.
Zhang, D. X. and Hewitt, G. M. (1996). Nuclear integrations: challenges for mitochondrial DNA markers. Trends Ecol Evol, 11: 247–251, 10.1016/0169-5347(96)10031-8.
Zink, R. M., Rohwer, S., Andreev, A. V. and Dittmann, D. L. (1995). Trans-Beringia comparisons of mitochondrial DNA differentiation in birds. Condor, 97: 639–649.
Acknowledgements
We are extremely grateful to T. H. Cohn (Museum of Zoology, University of Michigan), V. R. Vickery and C.-C. Hsiung (Lyman Entomological Museum, McGill University), A. Bugrov (Novosibirsk State University), A. Guéguen (University of Rennes) and R. A. Nichols (University of London) for providing museum specimens. This research was funded by a grant (W. C.) and postgraduate scholarships (G. L.) from the Natural Sciences and Engineering Research Council of Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Litzenberger, G., Chapco, W. A molecular phylogeographic perspective on a fifty-year-old taxonomic issue in grasshopper systematics. Heredity 86, 54–59 (2001). https://doi.org/10.1046/j.1365-2540.2001.00806.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.2001.00806.x
Keywords
This article is cited by
-
Evolutionary history and colonization patterns of the wing dimorphic grasshopper Dichroplus vittatus in two Argentinean biomes
Scientific Reports (2022)
-
The phylogeny of the Orthoptera (Insecta) as deduced from mitogenomic gene sequences
Zoological Studies (2013)
-
Phylogeographic studies on natural populations of the South American fruit fly, Anastrepha fraterculus (Diptera: Tephritidae)
Genetica (2007)
-
Diversity of Boll Weevil Populations in South America: A Phylogeographic Approach
Genetica (2006)
