
Abstract
We obtained 282 base pairs of sequence for the mitochondrial control region of 70 individuals of Korean striped field mice Apodemus agrarius coreae and Apodemus agrarius chejuensis to determine the levels of genetic divergence between these morphologically distinct taxa. The DNA sequences showed more genetic diversity (π) in A. agrarius coreae (2.98%) than in A. agrarius chejuensis (1.86%). Our data do not support the current concept that the two morphotypes are different species, but phylogenetic analysis indicates that animals of A. agrarius coreae with large body size from Wan Island cluster with the large-bodied A. agrarius chejuensis, and should be included in that taxon. As currently accepted A. agrarius coreae is not strictly monophyletic, because the large-bodied samples cluster within the range of mitochondrial variation of A. agrarius chejuensis. The fact that the two morphotypes do not share mitochondrial haplotypes (χ2=66, P < 0.001) suggests that there is little gene flow between them. A molecular clock estimate suggests that the two subspecies might have been isolated at the time of separation of the islands from the mainland.
Similar content being viewed by others

Introduction
The striped field mice, Apodemus agrarius Pallas 1771, inhabit an area from the western part of Germany to Korea and are a distinct species regarded as the member of the subgenus Apodemus (Thomas, 1906; Corbet, 1978). Jones & Johnson (1965) reported four subspecies in A. agrarius from Korea, which are mostly based on body colour and size: manchuricus in the extreme northern part of the peninsula, pallescens in the coastal lowlands of southern Korea, coreae throughout the major portion of the peninsula, and chejuensis on Cheju Island. More recently, Koh and coworkers (Koh, 1986, 1987, 1991; Koh & Yoo, 1992) reported that A. agrarius pallescens was a synonym of A. agrarius coreae, and two subspecies, A. agrarius chejuensis and A. agrarius coreae, were clearly distinct from each other both in their morphological characters and in mtDNA restriction fragment length polymorphisms (RFLP). From comparisons of morphometric characters, Koh (1991) also reached the conclusion that among eight subspecies of the striped field mice in Asia (subspecies agrarius, ningpoensis, pallidior, chevrieri, insulaemus, manchuricus, coreae and chejuensis), chejuensis is distinct (the largest in body size) and may be a different species. Although the mice from Wan Island are considered as A. agrarius coreae, they cluster with the large-bodied mice from Cheju Island in morphometric analyses (Koh, 1989). Despite the fact that the body size of small mammals is strongly affected by selection (Heaney, 1978), it is also evident that genetic divergence contributes to the body size difference (Roth & Klein, 1986).
Cheju Island was formed by a series of volcanic activities at the end of the Tertiary (Park, 1985). Thereafter, it was connected to the mainland during the Pleistocene and separated again about 10 000 years ago. Although many taxonomic studies have been conducted on Apodemus from the Korean peninsula, no direct assays of genetic divergence have been carried out.
DNA sequences have become the most frequently used tool for inferring phylogenetic history as they are the basic units of information encoded by organisms (Hillis et al., 1990). Mitochondrial (mt) DNA is a highly sensitive genetic marker suitable for studies of closely related taxa or populations of a variety of species because of its fast rate of evolution and characteristic maternal inheritance (Wilson et al., 1985). The mitochondrial control region has been attractive to evolutionary biologists for fine scale comparative studies because it is believed to be one of the fastest evolving segments in the animal mitochondrial genome (Dillon & Wright, 1993; Stewart & Baker, 1994; Lee et al., 1995). Using the polymerase chain reaction (PCR), the sequence of the first half of the control region can be readily obtained (Kocher et al., 1989).
In this study, we analysed DNA sequences of the first half of the mitochondrial control region from 70 individuals of A. agrarius coreae and A. agrarius chejuensis to investigate the level of genetic diversity within and between the two subspecies, and describe the phylogenetic relationships of these populations.
Materials and methods
Samples
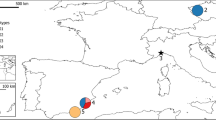
Seventy mice were trapped from five different localities in Korea (Fig. 1). A sample of A. peninsulae collected from the Korean mainland was used as an outgroup. Liver tissue was preserved in 100% ethanol and shipped to the University of New Hampshire for mtDNA sequencing and analysis. Voucher specimens have been deposited in the Department of Biology of Chungbuk National University, Korea.

Map of collection localities of Apodemus on the Korean Peninsula. The collection localities are boldface and underlined. The number of samples from each locality is as follows: Cheongju, 10; Mt. Jiri, 4; Haenam, 4; Wan Island, 41; Cheju Island, 10.
DNA extraction and PCR
Total genomic DNA was extracted from the tissues after first drying the samples to remove the ethanol. Liver tissue (0.01–0.03 g) was added to microtubes containing 500 μL of 5% Chelex and boiled for 15 min (Walsh et al., 1991). After a brief centrifugation, 10 μL of the supernatant was used as template for PCR.
Two primers, one based on the tRNA-Pro gene (5′-TTCCACCTCTAACTCCCAAAGCTAG-3′) and the other on the sequence of conserved block D (5′-CCTGAAGTAGGAACCAGATG-3′), were used for PCR amplification of the first half (5′-end) of the region (Kocher et al., 1989). For double-strand amplification, the following reagents were added to each microtube containing the genomic DNA: 5 μL 10× Taq buffer (0.67 M Tris, pH 8.8; 0.02 M MgCl2; 98 mM β-mercaptoethanol; and 0.1% Tween-20), 5 μL of both primers (10 μM), 1 μL 2.5 mM each dNTPs and 0.33 μL 5 unit Taq DNA polymerase. Each sample was brought up to 50 μL with sdH2O. The reaction was overlaid with 20–30 μL of mineral oil to prevent evaporation during thermal cycling. PCR conditions consisted of 30 thermal cycles of denaturation (93°C, 0.5 min), annealing (50°C, 1 min), and extension (72°C, 2 min) steps. After completion of all thermal cycles, the effectiveness of amplification was checked on 1% Seaplaque agarose gel. In all cases, a single, sharp band approximately 282 base pairs (bp) in length was observed and cut from the gel. The amplified DNA products were retrieved from the gel slice using agarase digestion (Sigma Chemical Co.).
Sequencing
We sequenced the amplified double-stranded DNA using the Taq DyeDeoxy Terminator cycle sequencing kit (Applied Biosystems) and an automated DNA sequencer (Applied Biosystems Inc., 373A). We sequenced using the same two primers as for PCR, and obtained sequences from both strands. After the cycle sequencing reaction, extra dye terminators were removed using spin columns containing 6% Sephadex. Finally, the entire sample was dried and resuspended in 4 μL of formamide buffer, denatured and loaded on the sequencer. All sequences have been deposited in Genbank under accession numbers U32276–U32304.
Sequence analysis
Analyses were performed with SeqEd (Applied Biosystems), ESEE (Cabot & Beckenbach, 1989), and the GCG package (ver. 7.0, Genetics Computer Group Inc.). We first aligned the D-loop sequences using the GCG program PILEUP with 1.0 and 0.3 for gap and gap length penalties, respectively. We then calculated gamma distances (Tamura & Nei, 1993) assuming a value of a=0.5. From these distances we obtained a phylogenetic tree by the Neighbour-Joining (NJ) method using the program MEGA (Kumar et al., 1993). The nucleotide diversity within the two conspecific populations was estimated following Nei (1987). Nucleotide diversity is defined as  , where πij is the proportion of different nucleotides between the ith and jth genotypes, and n is the total number of sequence comparisons, which is equal to {n(n − 1)/2}. The nucleotide diversity between two subspecies also can then be estimated by dA=dXY − (dX + dY)/2, where dXY is the averaged nucleotide diversity per site between haplotypes from the two subspecies, and dX and dY are the averaged nucleotide diversity per site within each subspecies. The significance of haplotypic variations between populations or geographical regions was calculated using the AMOVA (Excoffier et al., 1992) and Monte Carlo (McElroy et al., 1991) approaches.
, where πij is the proportion of different nucleotides between the ith and jth genotypes, and n is the total number of sequence comparisons, which is equal to {n(n − 1)/2}. The nucleotide diversity between two subspecies also can then be estimated by dA=dXY − (dX + dY)/2, where dXY is the averaged nucleotide diversity per site between haplotypes from the two subspecies, and dX and dY are the averaged nucleotide diversity per site within each subspecies. The significance of haplotypic variations between populations or geographical regions was calculated using the AMOVA (Excoffier et al., 1992) and Monte Carlo (McElroy et al., 1991) approaches.
Results
Sequence divergence
We obtained 282 bp of sequence for a portion of the control region (adjacent to tRNA-Pro) from 70 A. agrarius coreae and A. agrarius chejuensis. Forty-four unique haplotypes were found among the 70 individuals. Most of the base substitutions were found within the first 150 bp of the sequence. Among the 70 A. agrarius coreae and A. agrarius chejuensis, 57 sites out of 282 bp have experienced substitution. Within A. agrarius coreae and A. agrarius chejuensis, the number of variable sites was 39 (for the samples from the mainland only) and 22, respectively. More specifically, as seen in other mammals (Irwin et al., 1991), transitional changes outnumber the transversional substitutions. There are five unique base substitutions found between the two groups and all of them are transitional substitutions. One indel was observed between the two subspecies.
The nucleotide divergence among all samples of the two subspecies was 3.5%. Within each subspecies, A. agrarius coreae individuals showed about 1.6 times higher sequence variation than A. agrarius chejuensis individuals (2.98% vs. 1.86%, respectively). Nucleotide divergence between A. agrarius coreae and A. agrarius chejuensis was estimated excluding the samples from Wan Island, because they showed unique genotypes with sequence similarity closer to those from Cheju Island. The averaged nucleotide divergence between the two subspecies was 1.0%. The sequence divergence between samples from the mainland and Wan Island (1.59%) was slightly higher than that between the samples from the mainland and Cheju Island (1.0%). Despite the geographical distance between the two islands, the sequence divergence between individuals from Cheju and Wan islands (0.68%) was lower than that between these islands and the mainland (1.59% and 1.0%, respectively).
In the absence of gene flow between populations, the fast-evolving and maternally inherited mtDNA should give rise to nonoverlapping haplotype distributions in a short period of time (Watterson, 1985; Birky, 1991). We statistically compared the haplotype frequencies observed from each locality. First, we divided the localities of sampling into three regions (Mainland, Wan Island, and Cheju Island) for WINAMOVA (ver. 1.04), on the basis of geographical isolation. The fraction of haplotypic divergence among the three regions was higher (32%, P < 0.001) than the variance among populations within the regions (14%, P < 0.001) (Table 1). The results of Monte Carlo simulations are consistent with those of WINAMOVA. The samples from the three trap sites on the mainland (Fig. 1) did not exhibit significant haplotypic divergence (χ2=11.35, P < 0.0570). However, the haplotypic divergence among the three regions (Mainland, Wan, and Cheju islands) was significant (χ2=66, P < 0.001).
Phylogenetic analysis
The phylogenetic tree constructed by the Neighbour-Joining method in MEGA (Kumar et al., 1993) is presented in Fig. 2. In the analysis, the two subspecies are clustered as a monophyletic group with respect to the outgroup, Apodemus peninsulae. Clusters corresponding to Wan and Cheju islands are visible among a number of older mainland lineages. The samples from the two islands cluster into two groups, both distinguishable from the mainland samples. On the other hand, the branching order indicates that the genotypes of the inland samples collected from three different localities are randomly distributed with respect to the geographical location. The average gamma distance between A. agrarius coreae and A. peninsulae is 0.0501, and between A. agrarius chejuensis and A. peninsulae is 0.0659.

Neighbour-Joining tree of 70 samples of A. agrarius coreae rooted with A. peninsulae. The Tamura and Nei model (a=0.5) was used to obtain the distance matrix. The bootstrap values were obtained from 500 replications. Asterisks (* and **) represent the animals collected from Wan Island, but clustered with the mainland samples.
The distances between branches are relatively short throughout the tree, and bootstrapping (40%) does not strongly support the idea that A. agrarius chejuensis and the large-sized type from Wan Island are distinct from A. agrarius coreae. However the two morphotypes between Cheju Island and the mainland show no haplotype overlaps, indicating that little gene flow occurs between the Korean mainland and the coastal islands.
Discussion
Genetic divergence within the subspecies
These fast-evolving DNA sequences allowed us to quantify the amount of genetic variation between the two Korean subspecies of A. agrarius and to make comparisons with previous studies. Several intra- and interspecific variations in rodent mtDNA sequences have been reported. For instance, among three African mole rats (subspecies of Cryptomys hottentotus), the average sequence divergence was 15.8–20% and among six individuals of Georychus capensis, the divergence was about 12% (Honeycutt et al., 1987). In Apodemus species, the sequence diversity among Swedish A. flavicollis individuals was less than 1%, and nucleotide divergence between two different species (A. sylvaticus and A. flavicollis) was around 12% (Tegelstrom & Jaarola, 1989). The intraspecific variations between deer mice (Peromyscus maniculatus) from the California Channel Islands and the mainland, and between harvest mice (Reithrodontomys megalotis) collected from Santa Catalina Island and the mainland were up to 1% and 0.2%, respectively (Ashley & Willis, 1987; Ashley, 1989). From those previous studies, it becomes obvious that the levels of intraspecific divergence in rodents are highly variable, depending on the time of divergence and levels of gene flow.
The nucleotide diversity (1%) detected between these two A. agrarius subspecies is similar to the variation found in Swedish A. flavicollis. More sequence variation among individuals of A. agrarius coreae than among A. agrarius chejuensis (2.38% vs. 1.86%) may be attributable to either the larger population size of A. agrarius coreae, or the larger geographical range of this subspecies, or the fact that A. agrarius chejuensis are derived from a founder population. AMOVA and Monte Carlo tests indicate a significant genetic differentiation between the mainland and the two islands (χ2=66, P < 0.001). However, the three mainland localities share many haplotypes, suggesting that there is a significant amount of migration among them (χ2=11.35, P < 0.057).
A morphometric study of these populations found two distinct morphotypes (coreae, small-sized group; chejuensis, large-sized group) and suggested that they could be different species (Koh, 1986). However, the small genetic divergence (1%) and low bootstrap value (40%) revealed from this study suggest that the two morphotypes were isolated recently, but strongly supports that the large-sized A. coreae from Wan Island should be the same taxon as A. chejuensis. We suggest that the difference in body size between the subspecies may be enhanced by ecological factors like diet and predation, and will provide a good opportunity to investigate the causes for body size variation of insular mammals in East Asia.
Phylogeography and molecular time scale
Although the function of the first half of the animal mitochondrial control region is not yet clearly understood, no open reading frames within the control region have been found (Chang & Clayton, 1986). An a value of 0.5 is appropriate for calculating the gamma distance for phylogenetic analysis of noncoding regions like the D-loop in animal mtDNAs (Kumar et al., 1993). As shown in Fig. 2, the neighbour-joining phylogenetic tree based on this distance matrix indicates a geographically structured branching pattern. The geographical separations between mainland and islands leading to such genetic discontinuities between conspecific populations may be explained most easily by long-term barriers to gene flow.
The genotypes of individuals from Wan Island attract special interest. This island is located close to the mainland, but far from Cheju Island (Fig. 1). We might expect that the closer island has been isolated from the mainland for a shorter time (Wilcox, 1978). However, despite the indication of gene flow between Wan Island and the mainland (asterisked samples in Fig. 2), the nucleotide divergence between the samples from the two islands shows that they are much more closely related than between either island and the mainland (0.6% vs. 1–1.5%), supporting a morphometric study which showed that individuals from the two islands were clustered together as a large-sized morphotype (Koh, 1991). Ecological factors of these islands might act as a selective force for the large body size. However, it is also possible that the mice from Wan Island were introduced to Cheju Island. Prager et al. (1993) showed that the sequences of the mitochondrial control region were useful for tracking the migration route of house mice in East Holstein. Thus, more studies of the south coastal regions of the Korean peninsula and islands between Cheju and Wan Islands should be conducted to confirm whether the two genotypes coexist in some places and to reveal the migration pathway of the large-bodied mice.
The molecular data make it possible to estimate the time of divergence of the subspecies. It is generally estimated that the rate of nucleotide substitution in animal mitochondrial genomes is 2% per Myr based on primate RFLP studies (Brown et al., 1979). Irwin et al.’s (1991) analysis of mammalian mitochondrial cytochrome b gene sequences suggested that the rate of base substitution was similar to the previous estimation, 2.1% per Myr. Because of the relaxed functional constraint, the control region, especially the first half of the region, may evolve faster than the average rate of substitutions in the mitochondrial protein-coding genes (Brown, 1985). Wu & Li (1985) reported that the evolutionary rate of rodents was four to six times faster than that of primates, although there are uncertainties about the divergence times used for their calculation (Wilson et al., 1987). More recently the evolutionary rate of the shrew mitochondrial D-loop region was estimated to be 8.3%–14.3% (Stewart & Baker, 1994). Thus the mouse control region probably evolves at least 2% and possibly as much as 14% per Myr.
The sequence divergence of the control regions between the two subspecies is about 1%. It is hardly possible to estimate the exact divergence time of the two subspecies because of the variation in the sequence changes (2–14%/Myr). However, we can reasonably state that these two groups of mice have been isolated for less than a half million years, but longer than 7000 years. Cheju Island might have been isolated from the mainland around 10 000 years ago as other continental islands noted by Wilcox (1978). Therefore, it is quite possible that the two subspecies were separated at the time of the disconnection of the island from the mainland.
References
Ashley, M. V. (1989). Absence of differentiation in mitochondrial DNA of island and mainland harvest mice, Reithrodontomys megalotis. J Mammal, 70: 383–386.
Ashley, M. V. and Willis, C. (1987). Analysis of mitochondrial DNA polymorphism among Channel Island deer mice. Evolution, 41: 854–863.
Birky, C. W. (1991). Evolution and population genetics of organelle genes: mechanisms and models. In: Selander, R. K., Clark, A. G. and Whittam, T. S. (eds) Evolution at the Molecular Level. pp. 112–134. Sinauer Associates, Sunderland, MA.
Brown, W. M. (1985). The mitochondrial genome of animals. In: MacIntyre, R. J. (ed.) Molecular Evolutionary Genetics, pp. 95–130. Plenum, New York.
Brown, W. M., George, M. JR and Wilson, A. C. (1979). Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA, 76: 1967–1971.
Cabot, E. L. and Beckenbach, A. T. (1989). Simultaneous editing of multiple nucleic acid and protein sequences with Esee. Computer Applic Biosci, 5: 233–234.
Chang, D. D. and Clayton, D. A. (1986). Identification of primary transcriptional start sites of mouse mitochondrial DNA: Accurate in vitro initiation of both heavy- and light-strand transcripts. Mol Cell Biol, 6: 1446–1453.
Corbet, G. B. (1978). The Mammals of the Palaearctic Region: A Taxonomic Review. British Museum (Nat. Hist.), Cornell University Press, London and New York.
Dillon, M. C. and Wright, J. M. (1993). Nucleotide sequence of the D-loop region of the sperm whale (Physeter macrocephalus) mitochondrial genome. Mol Biol Evol, 10: 296–305.
Excoffier, L., Smouse, P. E. and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics, 131: 479–491.
Heaney, L. R. (1978). Island area and body size of insular mammals: evidence from the tri-colored squirrel (Callosciurus prevosti) of Southeast Asia. Evolution, 32: 29–44.
Hillis, D. M., Larson, A., Davis, S. K. and Zimmer, E. A. (1990). Nucleic acid III: Sequencing. In: Hillis, D. M. and Moritz, C. (eds) Molecular Systematics. pp. 318–369. Sinauer Associates, Sunderland, MA.
Honeycutt, R. L., Edwards, S. V., Nelson, K. and Nevo, E. (1987). Mitochondrial DNA variation and the phylogeny of African mole rats (Rodentia: Bathyergidae). Syst Zool, 36: 280–292.
Irwin, D. M., Kocher, T. D. and Wilson, A. C. (1991). Evolution of the cytochrome b gene of mammals. J Mol Evol, 32: 128–144.
Jones, J. K. JR and Johnson, D. H. (1965). Synopsis of the lagomorphs and rodents of Korea. Museum of Natural History (University of Kansas), 16: 357–407.
Kocher, T. D., Thomas, W. K., Meyer, A., Edwards, S. V., Paabo, S., Villablanca, F. X., Wilson, A. C. et al (1989). Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci USA, 86: 6196–6200.
Koh, H. S. (1986). Geographic variation of morphometric characters among three subspecies of striped field mice, Apodemus agrarius Pallas (Muridae, Rodentia) from Korea. Korean J Zool, 29: 272–282.
Koh, H. S. (1987). Systematic studies of Korean rodents: III. Morphometric and chromosomal analyses of striped field mice, Apodemus agrarius chejuensis Jones and Johnson, from Cheju-Do. Korean J Syst Zool, 3: 24–40.
Koh, H. S. (1989). Systematic studies of Korean rodents: V. Morphometric and chromosomal analyses in island populations of striped field mice (Apodemus agrarius coreae) in Southern coasts of the Korean peninsula. Korean J Syst Zool, 5: 1–12.
Koh, H. S. (1991). Multivariate analysis with morphometric characters of samples representing eight subspecies of striped field mice, Apodemus agrarius Pallas (Rodentia, Mammalia), in Asia: the taxonomic status of subspecies chejuensis at Cheju Island, Korea. Korean J Syst Zool, 7: 179–188.
Koh, H. S. and Yoo, B. S. (1992). Variation of mitochondrial DNA in two subspecies of striped field mice, Apodemus agrarius coreae and Apodemus agrarius chejuensis from Korea. Korean J Zool, 35: 332–338.
Kumar, S., Tamura, K. and Nei, M. (1993). Mega: Molecular evolutionary genetics analysis version 1.0. Pennsylvania State University, University Park, PA.
Lee, W. -J., Conroy, J., Howell, W. H. and Kocher, T. D. (1995). Structure and evolution of teleost mitochondrial control regions. J Mol Evol, 41: 54–66.
McElroy, D., Moran, P., Bermingham, E. and Kornfield, I. (1991). Reap: The Restriction Enzyme Analysis Package (version 4.0). J Hered, 83: 157–158.
Nei, M. (1987). Molecular Evolutionary Genetics. Columbia University Press, New York.
Park, D. W. (1985). Geology of coasts and mountains in Cheju-Do. Cheju-Do study II: 321–322. Cheju-Do, Korea.
Prager, E. M., Sage, R. D., Gyllensten, U., Thomas, W. K., Hubner, R., Jones, C. S. et al (1993). Mitochondrial DNA sequence diversity and the colonization of Scandinavia by house mice from Holstein. Biol J Linn Soc, 50: 85–122.
Roth, V. L. and Klein, M. S. (1986). Maternal effects on body size of large insular Peromyscus maniculatus: Evidence from embryo transfer experiment. J Mammal, 67: 37–45.
Stewart, D. T. and Baker, A. J. (1994). Pattern of sequence variation in the mitochondrial D-loop region of shrews. Mol Biol Evol, 11: 9–21.
Tamura, K. and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol, 10: 512–526.
Tegelström, H. and Jaarola, M. (1989). Genetic divergence in mitochondrial DNA between the wood mouse (Apodemus sylvaticus) and the yellow necked mouse (A. flavicollis). Hereditas, 111: 49–60.
Thomas, O. (1906). The Duke of Bedford’s zoological exploration in eastern Asia: III. List of small mammals from Korea and Quelpart. Proc Zool Soc Lon, 18: 858–865.
Walsh, P. S., Metzger, D. A. and Higuchi, R. (1991). Chelex-100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques, 10: 506–513.
Watterson, G. A. (1985). The genetic divergence of two populations. Theor Pop Biol, 27: 298–317.
Wilcox, B. A. (1978). Supersaturated island faunas: a species–age relationship for lizards on post-Pleistocene land-bridge islands. Science, 199: 996–998.
Wilson, A. C., Cann, R. L., Carr, S. M., George, M., Gyllensten, U. B., Helm-Bychowski, K. M. et al (1985). Mitochondrial DNA and two perspectives on evolutionary genetics. Biol J Linn Soc, 26: 375–400.
Wilson, A. C., Ochman, H. and Prager, E. M. (1987). Molecular time scale for evolution. Trends Genet, 3: 241–247.
Wu, C. -I. and Li, W. -H. (1985). Evidence for higher rates of nucleotide substitution in rodents than in man. Proc Natl Acad Sci USA, 82: 1741–1745.
Acknowledgements
We thank W. Kilpatrick and P. Rosel for helpful discussion. This study was supported by a grant from the Korean Science and Engineering Foundation (no. 91-0500-12-01-3) to H.S.K.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Koh, H., Lee, WJ. & Kocher, T. The genetic relationships of two subspecies of striped field mice, Apodemus agrarius coreae and Apodemus agrarius chejuensis. Heredity 85, 30–36 (2000). https://doi.org/10.1046/j.1365-2540.2000.00723.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.2000.00723.x
