
Abstract
Multilocus electrophoresis analysis has been used to study the genetic structure of 18 populations of Aedes albopictus newly introduced to Italy, in comparison with two populations in the United States, four in Japan, and four in Indonesia. Allozyme analysis revealed that 15 out of the 18 studied loci were polymorphic among the 28 populations. No significant deviations from Hardy–Weinberg equilibrium were found at polymorphic loci. High genetic affinity was observed between the Italian populations and those from the United States and Japan. The analysis of variance in allele frequencies showed that variance among subpopulations accounted for most of the total variance, suggesting that isolation of the Italian populations is not related to distance. Analysis of linkage disequilibrium using Ohta’s method shows that the variance in the frequency of allele combinations could be explained by the action of the genetic drift which accompanies the establishment of new populations. The colonization process of Ae. albopictus in Italy is following a trend similar to that previously observed in the U.S.A., probably because both infestations derive from several successive introductions, each with large numbers of individuals.
Similar content being viewed by others
Introduction
Aedes albopictus (Skuse, 1894) is considered one of the main vectors of dengue and can also play an important role in the transmission of a number of human arboviruses (Mitchell, 1995). The species was first described from specimens collected in Calcutta (Skuse, 1894), is included in the scutellaris group, subgenus Stegomya, and gives its name to a subgroup of 12 species mainly exploiting tree holes in the forests of south-east Asia. Although tropical forests are considered to be its original habitat, the species has developed the capacity to exploit artificial environments with a level of adaptation similar to that of Ae. aegypti. Unlike Ae. aegypti, Ae. albopictus is capable of colonizing temperate areas permanently, by overwintering in the egg stage through a diapausing embryo mechanism induced by a short photoperiod experienced by the female (Hawley et al., 1989). The capability to colonize used tyres is at the basis of the recent rapid establishment in new geographical areas (Reiter, 1998). In the 1980s Ae. albopictus was introduced into the United States where it progressively expanded and colonized some 24 States (Knudsen, 1995). Established populations have been detected in Brazil (eight States), the Caribbean (Santo Domingo, Dominican Republic), Mexico (three States), Guatemala, Honduras, El Salvador, Nigeria and the Fiji Islands. The species has been detected and probably eradicated in other areas such as Bolivia, Barbados, Cuba, New Zealand, Australia and South Africa (Knudsen, 1995).
In Europe, the first detection dates back to 1979 in Albania (Adhami & Murati, 1987). In Italy the species was first found in Genoa in September 1990 (Sabatini et al., 1990). From then on, its spread, mainly exploiting the national trade in used tyres, was very rapid and mosquito populations are now established in many regions. The effectiveness of efforts to stop the spread of the species has been hampered by the lack of regulations concerning storage and transportation of used tyres. We report a genetic study of the Italian populations of Ae. albopictus sampled 4–6 years after the species was first found in Italy. The aims of this research are to study the geographical origins of the Italian populations, to genetically characterize the structure of the populations, and to understand the reasons for the successful establishment of this species. Comparison with other colonization processes in different geographical areas will increase the genetic and ecological knowledge of this vector, and help us to understand the role of the colonization event in modelling population structure, and thereby to improve control strategies.
Materials and methods
Mosquito samples
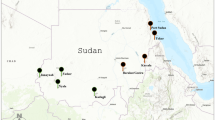
Aedes albopictus populations from 18 Italian localities were sampled over the 3-year period 1994–96 (Fig. 1). The collection mainly focused on larvae or adults, and when required ovitraps were used. Adults were immediately placed in insulated containers with dry ice and then transported to the laboratory, where they were stored in liquid nitrogen until examination. We also studied two samples of colonizing populations from the United States, Bradenton (19) and Lake Charles (20), and eight native populations: four from Japan, Choralcuji (21), Okinawa (22), Tanegashima (23) and Tnamaji (24), and four from Indonesia, Bali (25), Ujung Pandang I (26), Ujung Pandang II (27) and Toili (28).

Collection localities of the Italian populations of Aedes albopictus genetically analysed and their reference codes.
Electrophoresis analysis
Multilocus electrophoresis was carried out on single individuals. Homogenates were absorbed into 3 × 5 mm pieces of chromatography paper (Whatman 3 MM) and placed on 10% starch gel trays. Standard horizontal electrophoresis was carried out at 7–8 V/cm for 4–6 h at 5°C. We could analyse up to 15 enzyme systems for each mosquito, encoded by 19 putative loci (Table 1). Isozymes were numbered in order of decreasing mobility from the most anodal; allozymes were named numerically, according to their mobility relative to the most common allele (=100) in the sampled population.
Statistical analysis
We used the GENEPOP (version 3.1) software (Raymond & Rousset, 1995) to analyse population structure. Heterozygote deficits or excesses were tested using an exact test procedure (Rousset & Raymond, 1995). As measures of genetic variability, the number of alleles per locus, the percentage of polymorphic loci and the unbiased heterozygosity were calculated using the program BIOSYS-1 (Swofford & Selander, 1981). Unbiased estimates of F-statistics were calculated with FSTAT (Goudet, 1995) according to the formulae given in Weir & Cockerham (1984). Standard deviations for single-locus estimates were calculated by jackknifing over populations, and the 95% confidence limits of multilocus estimates were established using a bootstrap procedure over loci (Weir, 1990). Genotypic associations between each pair of loci were tested for each population by a probability test as described by Raymond & Rousset (1995), and over the whole data set by Ohta’s variance analysis (Ohta, 1982) using the LINKDOS program (Garnier-Gere & Dillmann, 1992). Genetic distance values between populations were estimated from allele frequencies using formulae by Rogers (1972), modified by Wright (1978). For genetic relationships between populations a multidimensional scaling ordination (Guttman, 1968) was carried out with the SYSTAT program (Wilkinson & Leland, 1989) from Rogers’s values, DT; this index was chosen because it is a metric coefficient satisfying the triangle inequality.
Isolation by distance was analysed as described by Rousset (1997), computing the relationship between pairwise estimates of FST/(1 − FST) and logarithms of geographical distance. A possible positive relationship was tested with a Mantel test, using the Spearman rank correlation coefficient as a statistic (as available in GENEPOP version 3.1). Geographical distances between samples were defined the shortest measurements on the map.
Results
Population structure of Italian populations
A total of 699 mosquitoes were analysed for 19 loci. In some cases, genotypes were not recorded for technical reasons. Overall, a total of 7140 genotypes were available for statistical analysis. Allozyme analysis revealed that 14 enzyme loci (G-3-pdh, Ldh, Hbdh, Mdh-1, Mdhp-1, Idh-1, 6-Pgdh, Aat-1, Hk-1, Adk-1, Acph, Aco-1, Gpi and Pgm) out of the 19 studied were polymorphic among the tested populations. The highest levels of polymorphism were observed at Hbdh, Acph and Pgm. The remaining five loci (Mdh-2, Idh-2, Sod-1, Mdhp-2 and Aco-2) were found to be monomorphic in all samples. Data concerning frequencies and FIS estimates of the most polymorphic loci are available upon request. Most of the populations were in Hardy–Weinberg equilibrium. Some samples were collected from several sites that were only a few metres apart and small observed heterozygote deficits may be an indication of a Wahlund effect. All polymorphic loci contributed significantly (P < 0.001) to the differentiation over all the populations. Another feature of the allele frequency data is the presence of rare alleles such as G-3pdh112 found in population no. 10, Hbdh116 in population no. 15, Idh93 in population no. 9 and Hk9, in population no. 12.
Table 2 reports the values of genetic variability found in the studied samples. The most variable populations were Rezzato (7), Montebelluna (1), Maragnole (5) and Bancole (10) with He=0.221, 0.219, 0.210 and 0.202, respectively. The least variable populations were Lonato (9) and Portogruaro (2) with He=0.134 and 0.144, respectively. These differences could be caused by genetic drift and/or inbreeding phenomena probably associated with differences in population size. The differentiation among Italian populations at various geographical scales was calculated by the variance in allele frequencies among subpopulations within provinces (populations not more than 20 km apart) and provinces within regions. The variances at greater distances were very low and not significantly greater than zero (Table 3). We used a method deduced from the theory of isolation by distance (Rousset, 1997), involving the regression of FST/(1 − FST) estimates for pairs of populations on the logarithm of distance between the populations. High differentiation was again observed at short distances, but the trend of increasing differentiation with distance was not significant (Mantel test, P=0.17).
The analysis of genotypic associations between pairs of loci in each sample revealed six nonrandom associations in 91 locus-pair comparisons. The tests for linkage disequilibrium were significant at P < 0.001 for, Pgm–Hbdh, Pgm–Idh-1, Pgi–6Pgdh, Hbdh–6Pgdh, 6Pgdh–Acph, and Idh-1–Aco-1. We followed Ohta’s (1982) analysis, which decomposes the gametic associations observed in the whole data set (DIT) into four indices, to discriminate the within population component (DIS and D′IS) from that between populations (DST and D′ST). For all pairs of loci, DIS < DST and D′IS > D′ST, indicating that genetic drift rather than selection is responsible for the nonrandom genotypic association found among Italian populations.
Analysis of geographical origin
Allozyme analysis conducted on Indonesian and Japanese samples (native populations) and American samples (populations of recent introduction) revealed that 15 enzyme loci out of the 19 studied were polymorphic among the tested populations. The remaining four loci (Idh-2, Sod-1, Mdhp-2 and Aco-2) were found to be monomorphic in all samples. The only difference from the Italian populations is that Mdh-2 is polymorphic. Table 2 shows the genetic variability values for the studied populations. The values of heterozygosity (He) in the temperate native populations are not significantly higher than those of the Italian ones.
Genetic relationships among the populations studied are illustrated by multidimensional scaling ordination (Fig. 2). Italian populations cluster together with American and Japanese ones, whereas samples from Indonesia are widely apart. There is a great separation between one pair of Indonesian populations, 28 and 25, and the other, 27 and 26. This pattern reflects the genetic heterogeneity in allele frequencies observed at four loci, G-3-pdh, Idh-1, Acph and Pgm, in these native populations. A similar variation was also found by Black et al. (1988) at loci G-3-pdh, Idh-1 and Pgm in Malaysian and Bornean populations. The high genetic differentiation among these samples suggests a geographical isolation among islands throughout Indonesia. Further research is needed to understand if the different allele frequencies are maintained by low migration among populations or/and by the action of selective pressures.

Plot of the first two dimensions of a multidimensional scaling ordination (Guttman, 1968), based on modified Rogers’ distance (DT; Rogers, 1972; Wright, 1978), showing genetic relationships among Italian, American, Japanese and Indonesian samples of Aedes albopictus.
Discussion
The used tyre trade with the United States has been identified as one of the factors responsible for the introduction of Ae. albopictus into Italy (Dalla Pozza et al., 1994). The high genetic affinity between the United States populations and the Italian populations support this hypothesis. Aedes albopictus must have been introduced into Italy by a large number of founder individuals, as demonstrated by the genetic variability values of Italian populations being similar to those of Japanese populations. Data on the international trade in used tyres reported by Reiter (1998) showed that Italy imported 44 687 used tyres from United States during 1989–94; 48 032 from Japan during 1988–95; 1550 from Taiwan during 1989–94. The newly introduced populations show a genetic structure quite similar to native Japanese populations, have been able to colonize new localities, increase their population sizes and compete with autocthonous species such as Culex pipiens (Celli et al., 1994). Although it is not possible to ascertain how many localities were colonized directly from abroad, following its arrival, the species spread, expanding the colonized area and increasing its population size. As a result, only 5 years after its first detection (although the species was probably introduced undetected before 1990) Ae. albopictus had rapidly expanded throughout northern and central Italy.
The colonizations of Ae. albopictus into Italy have followed a trend similar to those in the U.S. (Black et al., 1988), with large and repeated introductions. Its success is related to its biological and ecological characteristics, conferring adaptation to urban environments with temperate climates. In addition, however, the genetic characteristics of the colonizing groups seem to have played an important role in this success. The greater the size of the original colonizing group the higher is the probability that a population will become successfully established (McCauley, 1989).
In spite of the remarkable genetic homogeneity observed at a larger geographical scale, a high genetic differentiation was found among Italian populations, even those geographically close (only 10–20 km apart), suggesting the existence of local genetic drift. Analysis of linkage disequilibrium also suggested genetic drift, which can arise from a patchy distribution of the local populations, along with low dispersal and perhaps local founder events. For Ae. albopictus about 200 m is the maximum range of adult displacement from the breeding site during its life time (Hawley, 1988). This information is critical for future control. Severe restriction of gene exchange through habitat fragmentation could turn out to be a good strategy to control the expansion of this mosquito. Only continuous campaigns at source focused on reduction of the number of breeding localities, by creating barriers, could lead to a future loss of genetic variation, reducing the organism’s chances of further local expansion, and increasing our control capacity. Future research will evaluate how different control measures (strong or weak, larvicides or adulticides, with or without source reduction activity) may influence the evolution and expansion of new breeding populations, with the aim of planning a more efficient control programme.
References
Adhami, J. and Murati, N. (1987). Prani e mushkonjës Aedes albopictus në shqpëri. Revista Mjekësore. 1: 13–16.
Ayala, F. J., Powell, J. R., Tracey, M. L., Mourão, C. A. and Pérez-Salas, S. (1972). Enzyme variability in the Drosophila willistoni group. IV. Genic variation in natural population of Drosophila willistoni. Genetics, 70: 113–139.
Black, W. C. IV, Ferrari, J. A., Rai, K. S. and Sprenger, D. (1988). Breeding structure of a colonising species: Aedes albopictus (Skuse) in the United States. Heredity, 60: 173–181.
Brewer, G. J. and Sing, C. F. (1970). An Introduction to Isozyme Techniques. Academic Press, New York.
Celli, G., Bellini, R. and Carrieri, M. (1994). Survey on Aedes albopictus (Skuse) (Diptera: Culicidae) infestation in Desenzano del Garda (Brescia province-Italy). Boll Ist Ent ‘G Grandi’ Univ Bologna, 48: 211–217.
Dalla Pozza, G. L., Romi, R. and Severini, C. (1994). Source and spread of Aedes albopictus in the Veneto region of Italy. J Am Mosq Control Assoc, 10: 589–592.
Garnier-Gere, P. and Dillmann, C. (1992). A computer program for testing pairwise linkage disequilibrium in subdivided populations. J Hered, 83: 239–239.
Goudet, J. (1995). Fstat vers. 1.2: a computer program to calculate F-statistics. J Hered, 86: 485–486.
Guttman, L. A. (1968). A general nonmetric technique for finding the smallest coordinate space for a configuration of points. Psychometrika, 33: 469–506.
Harris, H. (1966). Enzyme polymorphism in man. Proc R Soc B, 169: 298–310.
Hawley, W. A. (1988). The biology of Aedes albopictus. J Am Mosq Control Assoc, 4 (Suppl. 1), 1–39.
Hawley, W. A., Punpini, C. B., Brady, R. H. and Craig, G. B. (1989). Overwintering survival of Ae. albopictus (Diptera Culicidae) eggs in Indiana. J Med Entomol, 26: 122–129.
Knudsen, A. B. (1995). Global distribution and continuing spread of Aedes albopictus. Parasitologia, 37: 91–97.
Mccauley, D. E. (1989). Extinction, colonization, and population structure: a study of milkweed beetle. Am Nat, 134: 365–376.
Menken, S. B. J. (1982). Enzymatic characterization of nine endoparasite species of small ermine moths (Yponomeutidae). Experientia, 38: 1461–1462.
Mitchell, C. J. (1995). Geographic spread of Aedes albopictus and potential for involvement in arbovirus cycles in the Mediterranean basin. J Vector Ecol, 20: 44–58.
Ohta, T. (1982). Linkage disequilibrium due to random genetic drift in finite populations. Proc Natl Acad Sci USA, 79: 1940–1949.
Poulik, M. D. (1957). Starch gel electrophoresis in a discontinuous system of buffer. Nature, 180: 1477–1477.
Raymond, M. and Rousset, F. (1995). GENEPOP (version 1.2), population genetics software for exact tests and ecumenicism. J Hered, 86: 248–249.
Reiter, P. (1998). Aedes albopictus and the world trade in used tyres, 1988–95: the shape of things to come?. J Am Mosq Control Assoc, 14: 83–94.
Rogers, J. S. (1972). Measures of genetic similarity and genetic distance. Studies in Genetics VII, University of Texas Publ, 7213: 145–153.
Rousset, F. (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics, 145: 1219–1228.
Rousset, F. and Raymond, M. (1995). Testing heterozygote excess and deficiency. Genetics, 140: 1413–1419.
Sabatini, A., Raineri, V., Trovato, G. and Coluzzi, M. (1990). Aedes albopictus in Italia e possibile diffusione della specie nell’area mediterranea. Parasitologia, 32: 301–304.
Selander, R. R., Smith, M. H., Yang, S. Y., Johnson, E. W. and Gentry, J. B. (1971). Biochemical polymorphism in the genus Peromyscus. Variation of the old-field mouse (Peromyscus polionotus). Studies in Genetics, University of Texas Publ, 7103: 49–90.
Shaw, C. and Prasad, R. (1970). Starch gel electrophoresis of enzymes: a compilation of recipes. Biochem Genet, 54: 297–320.
Skuse, F. A. A. (1894). The banded mosquito of Bengal. Indian Mus Notes, 30: 20–20.
Swofford, D. L. and Selander, R. R. (1981). BIOSYS-1: a FORTRAN program for the comprehensive analysis of electrophoretic data in population genetics and systematics. J Hered, 72: 281–283.
Weir, B. S. (1990). Intraspecific differentiation. In: Hillis, D. M. and Moritz, C. (eds) Molecular Systematics, pp. 373–470. Sinauer Associates, Sunderland, MA.
Weir, B. S. and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution, 38: 1358–1370.
Wilkinson, L. and Leland, J. (1989). SYSTAT: the System for Statistics. Systat Inc., Evanston, IL.
Wright, S. (1978). Evolution and the Genetics of Populations, vol. 4: Variability Within and Among Natural Populations. University of Chicago Press, Chicago, IL.
Acknowledgements
We are grateful to Prof. L. Bullini for his comments; two anonymous referees for their valuable contributions and suggestions; Dr M. Proietti, Dr V. Della Rosa and Dr L. Nini for their invaluable cooperation; Dr C.J. Mitchell, Prof. G. O’Meara, Dr M. Mogi, Dr A. Benedetti, Dr A. Bulgarelli, Dr S. Carasi, Dr A. Gasparetto, Dr V. Raineri, Dr M.G. Scialoia and Dr R. Zamburlini for kindly providing or helping in providing population samples. The research was carried out with grants from the Ministero dell’ Università e della Ricerca Scientifica e Tecnologica (40%).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Urbanelli, S., Bellini, R., Carrieri, M. et al. Population structure of Aedes albopictus (Skuse): the mosquito which is colonizing Mediterranean countries. Heredity 84, 331–337 (2000). https://doi.org/10.1046/j.1365-2540.2000.00676.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.2000.00676.x
Keywords
This article is cited by
-
Estimates of the dispersal of Chikungunya virus infected Aedes albopictus (Diptera: Culicidae) females from the retrospective spatio-temporal analysis of human cases during the 2007 Chikungunya outbreak in the Emilia-Romagna Region, Italy
International Journal of Tropical Insect Science (2022)
-
Transcriptional variation of sensory-related genes in natural populations of Aedes albopictus
BMC Genomics (2020)
-
High genetic diversity but no geographical structure of Aedes albopictus populations in Réunion Island
Parasites & Vectors (2019)
-
Population genetics of Aedes albopictus (Diptera: Culicidae) in its native range in Lao People’s Democratic Republic
Parasites & Vectors (2019)
-
Estimating the risk of arbovirus transmission in Southern Europe using vector competence data
Scientific Reports (2019)
