
Abstract
The genetic structure of metapopulations offers insights into the genetic consequences of local extinction and recolonization. We studied allozyme variation in rock pool metapopulations of two species of waterfleas (Daphnia) with the aim to understand how these dynamics influence genetic differentiation. We screened 138 populations of D. magna and 65 populations of D. longispina from an area in the archipelago of southern Finland. The pools from which they were sampled are separated by distances between 1.5 and 4710 m and located on a total of 38 islands. The genetic population structure of the two species was strikingly similar, consistent with their similar metapopulation ecology. The mean FPT value (differentiation among pools with respect to the total metapopulation) was 0.55 and a hierarchical analysis showed that genetic differentiation was strong (>0.25) among pools within islands as well as among whole islands. Within islands, pairwise genetic differentiation increased with geographic distance, indicating isolation by distance due to spatially limited dispersal. Previous studies have shown strong founder events occurring during colonization in our metapopulation. We suggest that the genetic population structure in the studied metapopulations is largely explained by three consequences of these founder events: (i) strong drift during colonization, (ii) local inbreeding, which results in hybrid vigour and increased effective migration rates after subsequent immigration, and (iii) effects of selection through hitchhiking of neutral genes with linked loci under selection.
Similar content being viewed by others


Introduction
Metapopulations describe populations structured into interconnected demes with local turnover dynamics of extinction and recolonization. Population turnover can have a strong impact on the distribution of genetic diversity within and among demes and thus on the evolution of populations (Slatkin, 1977; Wade and McCauley, 1988). This is because turnover often leads to genetic bottlenecks during colonization (‘founder events’) and because extinction limits the lifetime of individual demes and therefore the time during which subsequent gene flow can equalize allele frequencies. Therefore, in most cases, turnover is expected to increase genetic differentiation compared to similarly structured populations without turnover (Whitlock and McCauley, 1990) and this effect is most pronounced when the number of colonizers is small, when local populations are large, and when migration rates are low and spatially restricted (Whitlock and McCauley, 1990; Whitlock, 1992a).
Another important consequence of founder events is local inbreeding (‘drift load’), which in some situations leads to hybrid vigour after immigration (‘genetic rescue effect’; Whitlock et al, 2000; Ebert et al, 2002; Saccheri and Brakefield, 2002). Hybrid vigour increases effective migration rate because it confers a selective advantage to immigrant genes (Ingvarsson and Whitlock, 2000; Ebert et al, 2002). This process, in combination with increased drift during colonization, may contribute to a genetic population structure, in which young populations have a lower local genetic diversity and are more differentiated from each other than older populations (Whitlock, 1992a; McCauley et al, 1995; Nurnberger and Harrison, 1995; Giles and Goudet, 1997; Ingvarsson et al, 1997; Roach et al, 2001; Haag et al, 2005).
We have previously found an increase of local genetic diversity and a decrease of genetic differentiation with population age in two coexisting metapopulations of Daphnia species in small freshwater pools (Haag et al, 2005). Furthermore, in one of the species, hybrid vigour after immigration was confirmed experimentally (Ebert et al, 2002). In the present study, we carried out a detailed analysis of the spatial structure of genetic variation in these two metapopulations, because the geographic scale at which migration takes place may influence both genetic drift during colonization and selection for immigrant genes due to hybrid vigour. If migration is spatially restricted, genetic drift and consequently genetic differentiation are increased because colonizers of a given pool are likely to be genetically more similar to each other than under the island model of migration, where colonizers are randomly chosen from the whole metapopulation (Kimura and Weiss, 1964; Neigel, 1997). On the other hand, if migration is spatially restricted, immigrants are also likely to be genetically more similar to the resident population than under the island model. As a consequence, deleterious mutations are less likely to be masked in hybrids, causing hybrid vigour to be less strong for short-distance migrants than for long-distance migrants.
To investigate the spatial genetic population structure, we first assessed the hierarchical population structure of two sympatric Daphnia metapopulations, which are organized on three hierarchical levels: individuals are nested within pools, pools are nested within islands, and islands are nested within the whole metapopulation. In such a system, migration may take place predominantly within islands and only rarely between islands. This would also imply that the ecological dynamics of extinction and recolonization may take place more or less independently on each island, that is, each island would represent a small metapopulation of its own. Second, we assessed patterns of isolation by distance (Wright, 1943, 1946; Slatkin, 1993; Rousset, 1997) both within islands and among islands. Isolation by distance can be caused by spatially restricted migration and is assumed in most ecological metapopulation models (Brown and Kodric-Brown, 1977; Hanski, 1999). Finally, we compared the spatial genetic population structure of the two species, D. magna and D. longispina, which coexist in our study area and may co-occur even within the same pool (Pajunen, 1986; Pajunen and Pajunen, 2003). It has been proposed that the mechanism allowing this coexistence is that D. longispina is a better competitor, whereas D. magna is a better disperser (Hanski and Ranta, 1983). This would suggest that D. magna should be able to survive in places with larger interpool distances and should, for a given distance, show less genetic differentiation than D. longispina.
We sampled a high number of populations (138 in D. magna and 65 in D. longispina) and used allozymes to assess the spatial genetic population structure of the two species. Sampling of a high number of populations was crucial to achieve high power for the analysis of the hierarchical population structure and of isolation by distance.
Materials and methods
The study system
We investigated the genetic populations structure in metapopulations of the filter-feeding freshwater crustaceans D. magna and D. longispina (Cladocera). We studied D. magna and D. longispina that live in rock pools in the archipelago of southern Finland. Rock pools are small water-filled depressions in the bare rock and are often found on islands along the Baltic Sea coast. Daphnia rock pool populations form metapopulation systems, with discrete habitat patches and frequent local extinction and recolonization (Ranta, 1979; Hanski and Ranta, 1983; Pajunen, 1986; Bengtsson, 1989; Ebert et al, 2001, 2002; Pajunen and Pajunen, 2003). In our study area, the average proportion of pools containing D. magna or D. longispina populations in a given year is 18% for D. magna and 17% for D. longispina and the yearly extinction probability is 17 and 16%, respectively. The number of extinctions is, on average, balanced by the number of new colonizations (Pajunen, 1986; Pajunen and Pajunen, 2003). Even though the two species have slightly different habitat preferences (the pools vary in size, salinity, humic acid content, pH, calcium concentration, etc; Ranta, 1979), they often occur together (Pajunen, 1986). The two species have thus very similar metapopulation ecologies.
Both species reproduce by cyclical parthenogenesis, in which phases of asexual reproduction are intermitted by sexual reproduction. In our study area, Daphnia survive the winter as resting eggs. In spring, only females hatch and start to reproduce asexually. Later in the season, environmental sex determination leads to the clonal production of males (Hebert and Ward, 1972; Hobaek and Larsson, 1990), and subsequent sexual reproduction leads to the formation of resting eggs, which are needed to survive the winter. As resting eggs can only be produced sexually, each hatchling female is the founder of a genetically unique clone. The number of hatchlings in spring is hundreds to thousands and local populations often contain tens of thousands of individuals (C Haag, unpublished results). The length of an asexual generation (hatching to first reproduction) is 10–20 days and there are about 8–12 asexual generations during the season (ca. 5 months in our study area). Resting eggs are also the main dispersal stage, carried passively by wind, water, and birds. Owing to the possibility of clonal reproduction, a single hatchling female from a blown-in resting egg may found a new population, consisting of genetically identical individuals. In such uniform populations, all resting eggs are produced by within-clone mating, which is genetically equivalent to selfing.
The study area and sampling design
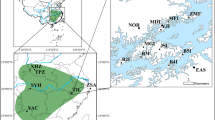
Our study area is in the archipelago of southern Finland at Tvrminne on the Hanko peninsula (5950′N, 2315′E; Figure 1), where we collected samples of D. magna between 5 June and 23 August 1998, and samples of D. longispina between 22 May and 28 September 1999. We divided the study area in two categories (Figure 1). In the core area consisting of 13 islands and a total of 507 pools, we sampled all inhabited pools (19% for D. magna and 13% for D. longispina). In the outer area, which forms a belt around the core area and contains more than 60 islands, we sampled one to four (usually two) populations per island from 23 islands for D. magna (total 63 pools) and from seven islands for D. longispina (total 17 pools). We included only isolated pools, excluding pools receiving water from other pools because they cannot be considered to be independent. Overall, we sampled a total of 138 populations on 36 islands for D. magna and 65 populations on 15 islands for D. longispina, separated by distances of 1.5–8938 and of 6–4710 m, respectively. The geographic location of each pool was recorded using a differential global positioning system (accuracy to the nearest meter). A description of all population parameters used in this study is given in Table 1.

Map of the study area on the Baltic coast of Southern Finland. Daphnia were sampled from islands shown in black (M: D. magna, L: D. longispina). The polygon surrounds islands of the core area, where all existing populations were sampled. To our knowledge, there are no populations of either of the two species present on the mainland (dashed) within 20 km, but there are populations on islands not included in the study. The cross indicates the location of Tvrminne Zoological Station at 5950′N and 2315′E.
Collection of allozyme data
Samples were collected with a hand net, with equal effort in all parts of a pool. Samples were brought to the laboratory, where they were kept alive at 12C and at low density to prevent selective mortality. Random samples of usually 22 individuals (range: 8–22 in D. magna; 20–22 in D. longispina) were screened for allozyme polymorphism by cellulose acetate electrophoresis (Hebert and Beaton, 1993). The following polymorphic loci were studied: aspartate amino transferase (Aat, enzyme commission number EC 2.6.1.1), fumarate hydratase (Fum, 4.2.1.2), glucose-6-phosphate isomerase (Gpi, 5.3.1.9), phosphoglucomutase (Pgm, 5.4.2.2), lactate dehydrogenase (Ldh, 1.1.1.27, only D. magna), malic enzyme (Me, 1.1.1.40, only D. longispina), and a peptidase locus with leucyleglycine as dipeptide substrate (Pep, 3.4.11, only D. longispina).
Data analysis
To analyse the genetic composition of populations, we estimated allele number, allelic richness (El Mousadik and Petit, 1996), and gene diversity (Nei, 1973) for each locus and for each population, using the program FSTAT (Goudet, 2001).
We assessed genetic structure by means of F-statistics (Weir and Cockerham, 1984). We use the subscripts I for individual, P for pool, L for island, and T for total. ‘Total’ is the total metapopulation, except for pairwise comparisons, where T is the total of a pair of pools or a pair of islands. We first used FSTAT to perform a classical two-level analysis to estimate FPT in order to assess the differentiation among pools compared to the total metapopulation (FPT measures the correlation of genes within pools relative to the total metapopulation). We obtained P-values for FPT by permutation tests (5000 permutation) and confidence intervals by bootstrapping over loci (15 000 repeats).
We used the program ARLEQUIN (Schneider et al, 2000) to run a three-level hierarchical analysis. We estimated FIP (the correlation of genes within individuals relative to pools), FPL (the correlation of genes within pools relative to islands), and FLT (the correlations of genes within islands relative to the total metapopulation). This analysis was carried out using the per locus AMOVA function and by averaging over loci by taking ratios of the sums of the variance components (for details, see Giles et al, 1998). P-values were obtained by 5000 permutations. Using FSTAT, we also tested for differences in FIP and FPL values among islands (5000 permutations). This was only done for the islands, where we had sampled all inhabited pools.
To test for isolation by distance, we analysed correlations between matrices of pairwise FPT values and matrices of pairwise geographic distances and tested their significance with Mantel tests (10000 randomizations), implemented in FSTAT. For islands with ≥4 samples, we also tested for isolation by distance within each island separately and used Fisher's method for combining probabilities (Sokal and Rohlf, 1981) to obtain a global test for isolation by distance within islands. Analyses of isolation by distance were carried out with untransformed FPT versus the logarithm (base 10) of geographic distance and with ranked FPT versus ranked geographical distance. Since several FPT values were equal to 1, we could not use FPT/(1−FPT) as suggested by Rousset (1997).
In D. magna 19% of pairwise FPT estimates were undefined, that is, both populations were fixed for the same, all-homozygous multi-locus genotype and hence expected total heterozygosity HT was zero. Undefined FPT values do not necessarily indicate a lack of differentiation, but can also reflect a lack of resolution due to low marker polymorphism. In D. longispina only 0.2% of pairwise estimates were undefined, and we set these missing estimates to zero, as would be done in Nei's (1987) genetic distance. However, in D. magna, this may have biased our FPT estimates downwards and, consequently, we analysed isolation by distance in D. magna by estimating Pearson's correlation coefficient and Spearman's coefficient of rank correlation between FPT and the logarithm (base 10) of geographic distance for a data set containing all pairs of pools with defined FPT values. As these pairs are subsets of matrices, treating them as independent samples would lead to pseudo-replication. However, a Mantel test, which would correct for this, could not be applied because of the missing values. We therefore evaluated the significance of these correlation coefficients as for independent samples, but using a corrected sample size Ncorr, which was estimated as the number of pools minus one (one less than the number of rows in the matrix), which is the number of independent distance estimates.
To test for within-island isolation by distance, we confined the data set to pairwise comparisons within islands (ie, all pairs with both pools on the same island) and estimated Ncorr as the number of pools minus the number of islands. This was again equal to the number of independent distance estimates because on each island the number of independent distances equals one less than the number of pools. We evaluated this method using the data set of D. longispina, for which we had obtained P-values for each of eight islands as well as for the overall data set with Mantel tests. We found that Mantel tests yielded in each case lower P-values than our tests based on Ncorr, and concluded that the latter method is more conservative.
To test for isolation by distance for gene pools of whole islands, we analysed Mantel correlations between pairwise FLT and geographic distance estimated from the logarithm (base 10) of distance between island centres. For this analysis, we excluded monomorphic islands, that is, islands on which only one multi-locus genotype was found.
We also tested whether the average geographic distance between pairs of pools differed between the two species. For this, we used a similar reasoning as explained above for correlations between incomplete matrices. We used a classical t-test, but estimated degrees of freedom and standard errors for this test from the number of pools in each species, rather than from the number of pairwise distances. This analysis was only carried out for the core area, where all existing populations were sampled.
Results
Allele number, allelic richness, and gene diversity for each locus are given in Table 2. Average within-pool gene diversities were less than half of the total gene diversities, and in no pool all alleles were found (Table 1). The metapopulations of both Daphnia species showed a strong genetic differentiation. The overall differentiation of pools compared to the metapopulation (FPT) was 0.56 for D. magna (bootstrapped 95% confidence limits: 0.49–0.59) and 0.56 for D. longispina (0.41–0.77). The results of the hierarchical analysis are given in Table 3. They show that there was a strong differentiation among pools within islands (D. magna: FPL=0.43, D. longispina: FPL=0.34), as well as among gene pools of whole islands (D. magna: FLT=0.24, D. longispina: FLT=0.34). Furthermore, on average, there was a highly significant excess of heterozygotes within populations of both species (FIP=−0.097 for D. magna and FIP=−0.22 for D. longispina, Table 3).
There was only little variation in within-island differentiation among different islands, indicating that the genetic population structure was similar on each of the islands. In D. magna, variation in FPL was marginally significant among islands (P=0.048), but not variation in FIP (P=0.56). In D. longispina, neither FIP nor FPL differed significantly among islands (FIP: P=0.67, FPL: P=0.28). Significant variation in FPL among islands in D. magna was mainly due to one island with a very low value.
Isolation by distance among all sampled pools was weak or absent (although significant for D. longispina, Table 4), but, within islands, pairwise FPT clearly increased with distance (Table 4, Figure 2). In contrast, for pairs of pools on different islands, there was no such correlation (Table 4, Figure 2). If undefined pairwise FPT values were set to zero, isolation by distance among all sampled pools became significant also in D. magna, but the relationship between pairwise FPT and geographic distance still levelled off after ca. 100 m (results not shown). The gene pools of whole islands (FLT) showed isolation by distance in D. magna (25 islands), but not in D. longispina (15 islands, Table 4, Figure 3).

Pairwise differentiation (FPT) between pools in relation to geographic distance for D. magna (a and b) and D. longispina (c and d). The upper two graphs show differentiation between pools on the same island, the lower two graphs show differentiation between pools on different islands. The fitted lines are linear least-square regressions between pairwise FPT and the logarithm (base 10) of distance. Distance is shown on a log 10 scale.

Pairwise differentiation (FLT) between islands in relation to geographic distance for D. magna (a) and D. longispina (b). The fitted lines are linear least-square regressions between pairwise FLT and the logarithm (base 10) of distance. Distance is shown on a log 10 scale.
The average geographic distance between pairs of pools in the core area was 911 m in D. magna and 916 m in D. longispina. The difference between the two species was not significant (t-test using log 10-transformed distances, t=−0.38, corrected df=124, P=0.70). Neither the geographic distances between pairs of pools within islands nor between pairs of pools between islands differed significantly between the two species, although within-island geographic distances were 27% shorter in D. magna (D. magna: 49 m, D. longispina: 67 m, t=−1.78, df=111, P=0.08).
Discussion
Explaining the high levels of genetic differentiation
In this study, we found high levels of among-population genetic differentiation in metapopulations of two co-occurring species of Daphnia, corroborating earlier evidence (Korpelainen, 1984; Haag et al, 2005) that rock pool metapopulations of Daphnia show strong genetic differentiation. These high levels of genetic differentiation, even among pools only a few meters apart, are likely to have multiple reasons. Yet, we believe that the very strong genetic bottlenecks during colonization represent one of the key factors. Rock pool populations of Daphnia are often founded by very few individuals, sometimes perhaps even by single individuals (Ebert et al, 2002; Haag et al, 2005). As dispersal is passive, it can be assumed that immigration occurs at a similar rate as colonization (ie, colonization is successful immigration into an empty pool). The strong reduction in population size during colonization leads to strong genetic drift, that is, founder events. In our system, founder events are expected to be particularly important because with high local population sizes and low migration rates, the approach to equilibrium after a bottleneck is slow and because local populations have only a limited lifetime due to high extinction rates (Crow and Aoki, 1984; Whitlock and McCauley, 1990; Whitlock, 1992b). Founder events are thus expected to lead to high genetic differentiation (Wade and McCauley, 1988), and this may even be accentuated by spatially restricted migration (Whitlock and McCauley, 1990; Pannell and Charlesworth, 1999).
Strong genetic drift during colonization coupled with a rapid subsequent increase in population size may also be the reason for strong genetic differentiation in other cyclical parthenogens (eg, Hebert, 1987; Lynch and Spitze, 1994; De Meester et al, 2002). In cyclical parthenogens, a few or even a single colonizer can rapidly give rise to enormous populations, whereas immigrants have to compete with the resident population and do not undergo this enormous increase in population size. Due to this, sampling effects on founder allele frequencies can persist for many generations (‘persistent founder effects’, Boileau et al, 1992; Berg and Lascoux, 2000; Palsson, 2000; De Meester et al, 2002; Gomez et al, 2002). Persisting founder effects may also cause correlations between observed differentiation and gene flow to be weak in cyclical parthenogens, which may be a reason for weak or noisy patterns of isolation by distance in cyclical parthenogens (eg, Innes, 1991; Lynch and Spitze, 1994; Vanoverbeke and De Meester, 1997).
Besides increased drift during colonization, genetic bottlenecks also lead to linkage disequilibrium, which may have non-neutral effects on genetic population structure. Linkage disequilibrium caused by genetic bottlenecks can persist for several generations and effects of background selection on linked loci may consequently be increased (Charlesworth et al, 1993). With background selection, neutral alleles change in frequency due to selection at linked loci. In Daphnia, this effect may be especially strong because of genomewide linkage during clonal selection (Vanoverbeke and De Meester, 1997; Berg and Lascoux, 2000; Palsson, 2001). Linkage with deleterious alleles can also lead to associative overdominance (Ohta, 1971). Associative overdominance tends to reduce genetic differentiation, whereas background selection tends to increase it (Charlesworth et al, 1997; Palsson and Pamilo, 1999; Pamilo et al, 1999) and currently it is unclear which of the two effects may be stronger in our Daphnia metapopulations.
Finally, some of our within-pool samples may have contained multiple copies of the same clones. This is especially likely for populations founded in the year of sampling and for populations sampled a relatively long time after hatching. Inclusion of multiple clonal copies can increase the overall genetic differentiation because of random associations of alleles with successful clones. This is sometimes overcome by including only one individual per multi-locus genotype in each population sample (Sunnucks et al, 1997), assuming that individuals with the same multi-locus genotype are clonal copies. In the present study, it is likely that this assumption was not fulfilled because the number of hatchlings (founders of unique clones) within the pool is typically large (>1000, CR Haag, unpublished results) and the vast majority of hatchlings could not be distinguished by our markers (ie, the number of multi-locus genotypes was much smaller than the number of hatchlings). Thus, exclusion of multiple genotypes would have strongly biased our estimates in favour of rare alleles. Furthermore, allele frequencies in cyclical parthenogens are also influenced by random associations of alleles with successful clones in the previous season and not only by the inclusion of multiple copies. The contribution of clonal selection to genetic differentiation in cyclical parthenogens is thus an inherent property of this breeding system.
Within-island and among-island genetic population structure
High levels of genetic differentiation were evident not only among single local populations but also among gene pools of whole islands. This suggests that gene flow between islands is limited compared to that within islands due to larger geographic distances between islands, or perhaps also because of potentially different dispersal mechanisms for within-island and between-island migration. Within islands there was a clear pattern of isolation by distance, indicating that gene flow was spatially restricted also within islands. Specifically, most colonizers and most immigrants are likely to originate from nearby pools on the same island. Together, these results suggest that the ecological dynamics of extinction and recolonization may act largely independently on each of the islands (‘semi-independent networks’; Hanski, 1998). Populations structured in this way offer excellent opportunities to study metapopulation processes, because of the possibility to use the islands as unit of replication, and thus to replicate metapopulations. Metapopulations with similar hierarchical genetic population structures have been found in a plant and a beetle species in other archipelago landscapes (Ingvarsson and Olsson, 1997; Giles et al, 1998).
At a larger geographic scale, covering many islands, there was no detectable (D. magna) or only weak (D. longispina) isolation by distance. The relationship between genetic differentiation and geographic distance appeared to level off after about 100 m. This observation may possibly be explained by long-distance migrants having a disproportionately large equalizing effect on gene frequencies among populations (Slatkin, 1985; Nichols and Hewitt, 1994) because long-distance migration may lead to extensive hybrid vigour and therefore substantially increase the effective migration rate (Ingvarsson and Whitlock, 2000; Whitlock et al, 2000). Short-distance migrants, on the other hand, are likely to immigrate into genetically similar populations and, consequently, hybrid vigour and its effect on gene flow may be less pronounced because homozygous deleterious alleles are less likely to be masked in the resulting hybrids. This mechanism would suggest that the pattern of isolation by distance on a local scale is largely influenced by drift, whereas selection becomes more important at a larger geographic scale. It is currently unknown whether the magnitude of hybrid vigour in Daphnia depends on dispersal distance, but in D. magna hybrid vigour after between-island dispersal increases the effective migration rates manifold (Ebert et al, 2002).
There are, however, alternative explanations for the lack of genetic differentiation at larger geographic scales. First, at larger geographic scales, levels of genetic differentiation may approach their maximum because as long as some variation is maintained within populations (HS>0), FPT is not expected to tend to 1, even in the absence of gene flow. Second, allozyme polymorphisms at the level of the metapopulation may be maintained by balancing selection. If balancing selection is weak, it may only be effective at large geographic scales because locally the effects of selection are overruled by genetic drift (that is, because the effective size of the metapopulation is much larger than the local effective population size).
Even within islands, geographic distance explained only about 10% of the variance in genetic differentiation. Patterns of isolation by distance in highly structured metapopulations are often highly scattered and, consequently, large samples are needed for its detection (eg, Nurnberger and Harrison, 1995; Giles and Goudet, 1997; Roach et al, 2001; Charbonnel et al, 2002; Massonnet et al, 2002). This is due to the highly stochastic effect of turnover on the distribution of genetic diversity so that observed correlations can be weak, even if migration rate is strictly negatively correlated with geographic distance (Whitlock, 1992a; Ingvarsson and Olsson, 1997). Furthermore, in some metapopulations, including the two studied Daphnia metapopulations, younger populations tend to be more strongly differentiated than older populations (Whitlock, 1992a; McCauley et al, 1995; Nurnberger and Harrison, 1995; Giles and Goudet, 1997; Ingvarsson et al, 1997; Roach et al, 2001; Haag et al, 2005). This can induce additional variation around the relationship between genetic and geographic distance. The same is true if environmental factors such as pool volume or salinity affect genetic differentiation. In an earlier study we have tested for such effects, finding increased genetic differentiation between smaller pools compared to larger pools (Haag et al, 2005). Other tested environmental variables did not appear to have a strong effect.
Heterozygote excess within populations
On average, there was an excess of heterozygotes within local populations of both species. This was mainly due to six D. magna populations and five D. longispina populations with a FIP of −1, that is, consisting only of heterozygous individuals. This situation may arise either in newly colonized populations that have been colonized by very few (or single) heterozygous individuals or after strong selection for heterozygous individuals, for instance, due to hybrid vigour after immigration (Ebert et al, 2002). Excluding these populations, overall FIP was nonsignificant for D. magna (average=−0.02, P=0.32), but still highly significant for D. longispina (average=−0.157, P<0.0001). A potential reason for this may be that D. longispina populations were sampled slightly later in the season, allowing clonal selection to act for a longer period. There was also substantial variation in FIP values among loci. This was partially also due to the populations consisting of only heterozygous individuals and partially due to small sample size at some loci, but had only little influence on genetic differentiation and isolation by distance.
Comparison of the two species
The genetic population structures of the two species were strikingly similar, even quantitatively. Taking into account that also geographic distances between demes and metapopulation parameters (extinction and colonization rates; Pajunen and Pajunen, 2003) were very similar in the two species, our results do not provide evidence for the hypothesis by Hanski and Ranta (1983) that D. magna is a better disperser (ie, has a higher migration rate) than D. longispina. In contrast, geographic distances among pools within islands tended to be lower and genetic differentiation somewhat higher in D. magna than in D. longispina. Overall, the similarities in metapopulation ecology and population genetic structure suggest that extinction and colonization in these two species are governed by similar processes (see also Pajunen and Pajunen, 2003). How the two species coexist in the same area remains therefore unresolved, although slight differences in habitat preferences are perhaps the most likely explanation (VI Pajunen, unpublished manuscript).
References
Bengtsson J (1989). Interspecific competition increases local extinction rate in a metapopulation system. Nature 340: 713–715.
Berg LM, Lascoux M (2000). Neutral genetic differentiation in an island model with cyclical parthenogenesis. J Evol Biol 13: 488–494.
Boileau MG, Hebert PDN, Schwartz SS (1992). Non-equilibrium gene frequency divergence: persistent founder effects in natural populations. J Evol Biol 5: 25–39.
Brown JH, Kodric-Brown A (1977). Turnover rates in insular biogeography – effect of immigration on extinction. Ecology 58: 445–449.
Charbonnel N, Angers B, Rastavonjizay R, Bremond P, Jarne P (2002). Evolutionary aspects of the metapopulation dynamics of Biomphalaria pfeifferi, the intermediate host of Schistosoma mansoni. J Evol Biol 15: 248–261.
Charlesworth B, Morgan MT, Charlesworth D (1993). The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303.
Charlesworth B, Nordborg M, Charlesworth D (1997). The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genet Res 70: 155–174.
Crow JF, Aoki K (1984). Group selection for a polygenic behavioural trait: estimating the degree of population subdivision. Proc Natl Acad Sci USA 81: 6073–6077.
De Meester L, Gomez A, Okamura B, Schwenk K (2002). The monopolization hypothesis and the dispersal-gene flow paradox in aquatic organisms. Acta Oecol 23: 121–135.
Ebert D, Haag C, Kirkpatrick M, Riek M, Hottinger JW, Pajunen VI (2002). A selective advantage to immigrant genes in a Daphnia metapopulation. Science 295: 485–488.
Ebert D, Hottinger JW, Pajunen VI (2001). Temporal and spatial dynamics of parasites in a Daphnia metapopulation: which factors explain parasite richness? Ecology 82: 3417–3434.
El Mousadik A, Petit RJ (1996). High level of genetic differentiation for allelic richness among populations of the argan tree (Argania spinosa (L.) Skeels) endemic to Morocco. Theor Appl Genet 92: 832–839.
Giles BE, Goudet J (1997). Genetic differentiation in Silene dioica metapopulations: estimation of spatiotemporal effects in a successional plant species. Am Nat 149: 507–526.
Giles BE, Lundqvist E, Goudet J (1998). Restricted gene flow and subpopulation differentiation in Silene dioica. Heredity 80: 715–723.
Gomez A, Adcock GJ, Lunt DH, Carvalho GR (2002). The interplay between colonization history and gene flow in passively dispersing zooplankton: microsatellite analysis of rotifer resting egg banks. J Evol Biol 15: 158–171.
Goudet J (2001). FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3) (http://www2.unil.ch/popgen/softwares/fstat.htm).
Haag CR, Riek M, Hottinger JW, Pajunen VI, Ebert D (2005). Genetic structure in metapopulations with subpopulations of known age. Genetics 170: 1809–1820.
Hanski I (1998). Metapopulation dynamics. Nature 396: 41–49.
Hanski I (1999). Metapopulation Ecology. Oxford University Press: Oxford.
Hanski I, Ranta E (1983). Coexistence in a patchy environment: three species of Daphnia in rock pools. J Anim Ecol 52: 263–279.
Hebert PDN (1987). Genotypic characteristics of the Cladocera. Hydrobiologia 145: 183–193.
Hebert PDN, Beaton MJ (1993). Methodologies for Allozyme Analysis Using Cellulose Acetate Electrophoresis, 2nd edn. Helena Laboratories: Beaumont.
Hebert PDN, Ward RD (1972). Inheritance during parthenogenesis in Daphnia magna. Genetics 71: 639–642.
Hobaek A, Larsson P (1990). Sex determination in Daphnia magna. Ecology 71: 2255–2268.
Ingvarsson PK, Olsson K (1997). Hierarchical genetic structure and effective population sizes in Phalacrus substriatus. Heredity 79: 153–161.
Ingvarsson PK, Olsson K, Ericson L (1997). Extinction–recolonization dynamics in the mycophagous beetle Phalacrus substriatus. Evolution 51: 187–195.
Ingvarsson PK, Whitlock MC (2000). Heterosis increases the effective migration rate. Proc R Soc London B 267: 1321–1326.
Innes DJ (1991). Geographic patterns of genetic differentiation among sexual populations of Daphnia pulex. Can J Zool 69: 995–1003.
Kimura M, Weiss GH (1964). The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49: 561–576.
Korpelainen H (1984). Genetic differentiation of Daphnia magna populations. Hereditas 101: 209–216.
Lynch M, Spitze K (1994). Evolutionary genetics of Daphnia. In: Real LA (ed) Ecological Genetics. Princeton University Press: Princeton, New Jersey. pp 109–128.
Massonnet B, Simon JC, Weisser WW (2002). Metapopulation structure of the specialized herbivore Macrosiphoniella tanacetaria (Homoptera, Aphididae). Mol Ecol 11: 2511–2521.
McCauley DE, Raveill J, Antonovics J (1995). Local founding events as determinants of genetic structure in a plant metapopulation. Heredity 75: 630–636.
Nei M (1973). Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70: 3321–3323.
Nei M (1987). Molecular Evolutionary Genetics. Columbia University Press: New York.
Neigel JE (1997). A comparison of alternative strategies for estimating gene flow from genetic markers. Annu Rev Ecol Syst 28: 105–128.
Nichols RA, Hewitt GM (1994). The genetic consequences of long distance dispersal during colonization. Heredity 72: 312–317.
Nurnberger B, Harrison RG (1995). Spatial population structure in the whirligig beetle Dineutus assimilis – evolutionary inferences based on mitochondrial DNA and field data. Evolution 49: 266–275.
Ohta T (1971). Associative overdominance caused by linked detrimental mutations. Genet Res 18: 277–286.
Pajunen VI (1986). Distributional dynamics of Daphnia species in a rock-pool environment. Ann Zool Fenn 23: 131–140.
Pajunen VI, Pajunen I (2003). Long-term dynamics in rock pool Daphnia metapopulations. Ecography 26: 731–738.
Palsson S (2000). Microsatellite variation in Daphnia pulex from both sides of the Baltic Sea. Mol Ecol 9: 1075–1088.
Palsson S (2001). The effects of deleterious mutations in cyclically parthenogenetic organisms. J Theor Biol 208: 201–214.
Palsson S, Pamilo P (1999). The effects of deleterious mutations on linked, neutral variation in small populations. Genetics 153: 475–483.
Pamilo P, Plsson S, Savolainen O (1999). Deleterious mutations can reduce differentiation in small, subdivided populations. Hereditas 130: 257–264.
Pannell JR, Charlesworth B (1999). Neutral genetic diversity in a metapopulation with recurrent local extinction and recolonization. Evolution 53: 664–676.
Ranta E (1979). Niche of Daphnia species in rock pools. Arch Hydrobiol 87: 205–223.
Roach JL, Stapp P, Van Horne B, Antolin MF (2001). Genetic structure of a metapopulation of black-tailed prairie dogs. J Mammal 82: 946–959.
Rousset F (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Saccheri IJ, Brakefield PM (2002). Rapid spread of immigrant genomes into inbred populations. Proc R Soc Lond B 269: 1073–1078.
Schneider S, Roessli D, Excoffier L (2000). Arlequin ver.2.000: A software for population genetics data analysis (http://anthro.unige.ch/arlequin/).
Slatkin M (1977). Gene flow and genetic drift in a species subject to frequent local extinctions. Theor Popul Biol 12: 253–262.
Slatkin M (1985). Gene flow in natural populations. Annu Rev Ecol Syst 16: 393–430.
Slatkin M (1993). Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47: 264–279.
Sokal RR, Rohlf FJ (1981). Biometry, 2nd edn. Freeman: New York.
Sunnucks P, England PR, Taylor AC, Hales DF (1997). Microsatellite and chromosome evolution of parthenogenetic sitobion aphids in Australia. Genetics 144: 747–756.
Vanoverbeke J, De Meester L (1997). Among-populational genetic differentiation in the cyclical parthenogen Daphnia magna (Crustacea, Anomopoda) and its relation to geographic distance and clonal diversity. Hydrobiologia 360: 135–142.
Wade MJ, McCauley DE (1988). Extinction and recolonization: their effects on the genetic differentiation of local populations. Evolution 42: 995–1005.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Whitlock MC (1992a). Nonequilibrium population structure in forked fungus beetles: extinction, colonization, and the genetic variance among populations. Am Nat 139: 952–970.
Whitlock MC (1992b). Temporal fluctuations in demographic parameters and the genetic variance among populations. Evolution 46: 608–616.
Whitlock MC, Ingvarsson PK, Hatfield T (2000). Local drift load and the heterosis of interconnected populations. Heredity 84: 452–457.
Whitlock MC, McCauley DE (1990). Some population genetic consequences of colony formation and extinction – Genetic correlations within founding groups. Evolution 44: 1717–1724.
Wright S (1943). Isolation by distance. Genetics 28: 114–138.
Wright S (1946). Isolation by distance under diverse systems of mating. Genetics 31: 39–59.
Acknowledgements
We thank B Gimelli, I Pajunen, and the staff of Tvrminne Zoological Station for support and technical assistance. We profited from comments by G Goudet, P Jarne, T Kawecki, C Liautard, C Pampoulie, P Sepp and two reviewers. The work was supported by the Swiss National Fund, the Handschin-Stiftung, the Dr Max Husmann-Stiftung, the Josef & Olga Tomcsik-Stiftung, the Werenfels-Fonds (Freiwillige Akademische Gesellschaft Basel), and by the Roche Research Foundation. This is part of project nr 97524006 at Tvrminne Zoological Station.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haag, C., Riek, M., Hottinger, J. et al. Founder events as determinants of within-island and among-island genetic structure of Daphnia metapopulations. Heredity 96, 150–158 (2006). https://doi.org/10.1038/sj.hdy.6800774
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800774
Keywords
This article is cited by
-
Large effective size as determinant of population persistence in Anostraca (Crustacea: Branchiopoda)
Conservation Genetics (2023)
-
The contrasting genetic patterns of two sympatric flying fox species from the Comoros and the implications for conservation
Conservation Genetics (2018)
-
Isolation mediates persistent founder effects on zooplankton colonisation in new temporary ponds
Scientific Reports (2017)
-
Dormancy and dispersal as mediators of zooplankton population and community dynamics along a hydrological disturbance gradient in inland temporary pools
Hydrobiologia (2017)
-
Stress tolerance and population stability of rock pool Daphnia in relation to local conditions and population isolation
Hydrobiologia (2015)
