
Abstract
Biogeographic boundaries are characterised by distinct faunal and floral assemblages restricted on either side, but patterns among groups of taxa often vary and may not be discrete. Historical biogeography as a consequence, while providing crucial insights into the relationship between biological diversity and earth history, has some limitations. Patterns of intraspecific molecular variation, however, may show unambiguous evidence for such historical divides, and can be used to test competing biogeographic hypotheses (often based on the dispersal-vicariance debate). Here, we utilise this method to test the hypothesis that a major biogeographic transition zone between the Sundaic and Indochinese biotas, located just north of the Isthmus of Kra in SE Asia, is the result of Neogene marine transgressions that breached the Isthmus in two locations for prolonged periods of time (>1 million year duration). Phylogeographic analyses of a freshwater decapod crustacean, the giant freshwater prawn Macrobrachium rosenbergii, strongly supports the historical existence of the more northerly postulated seaway. Results presented here highlight the power of utilising intraspecific molecular variation in testing biogeographical hypotheses.
Similar content being viewed by others


Introduction
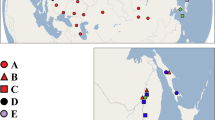
The biogeographic province of Sundaland (land on the Sunda Shelf south of the Isthmus of Kra) was greatly affected by eustatic sea-level changes for much of the Tertiary, including the well-documented flooding of the Sunda Shelf (Hanebuth et al, 2000). The region is bordered to the east by what is probably the most well-known biogeographic boundary recognised today, Wallace's Line. A second but lesser-known biogeographic boundary occurs at the transition zone between the Sundaic and Indochinese biotas (sensu Woodruff, 2003) in the vicinity of the Isthmus of Kra (Figure 1), with distinct assemblages of amphibians (Inger, 1966; see review by Inger and Voris, 2001), reptiles (Inger and Voris, 2001), birds (Hughes et al, 2003), mammals (Corbett and Hill, 1992), insects (Corbet, 1941) and plants (Ridder-Numan, 1998; Denduangboripant and Cronk, 2000) limited to varying degrees either side of this barrier. Recently, it has been hypothesised that marine transgressions may have produced this pattern (Woodruff, 2003); specifically, that Miocene- (24–23 Mya) and Pliocene-era (5.5–4.5 Mya) high sea-level stands resulted in two seaways that dissected the Thai-Malay Peninsula (Figure 1; Woodruff, 2003), for durations in excess of 1 million years. This hypothesis is based on an extensive review of both biological and geological evidence (Woodruff, 2003), and received strong support specifically from past sea-level highstands evident from the Vail global eustatic curve (Vail and Hardenbol, 1979) and the oxygen isotope curve (see Woodruff, 2003).

Study region and relationships among M. rosenbergii COI mtDNA haplotypes. Solid lines indicate approximate width of proposed Isthmus of Kra Seaways (Woodruff, 2003). Sampling sites labelled a–k (see Table 1 for details). The size of the circles and rectangle indicate relative frequencies of the haplotypes (Table 2). The single hatched circle in the southern clade indicates a ‘southern haplotype’ collected from a northern site (Kraburi River; site c).
If seaways had divided the Thai-Malay Peninsula in the past creating an archipelago for a significant period of time (>1 MY), this should be evident in the intraspecific molecular ‘signatures’ of organisms sampled from either side of this barrier, relative to their dispersal potential and the nature (width, throughflow volume, etc) of the seaway. Such studies on terrestrial (Hoffman and Baker, 2003; Zeh et al, 2003) and marine taxa (Knowlton et al, 1993; Collins et al, 1996; Bermingham et al, 1997; Knowlton and Weigt, 1998; Tringali et al, 1999; Marko, 2002 and others) from the Isthmus of Panama have provided a wealth of information on the role that vicariance can play in shaping genetic divergence among populations, and ultimately, in speciation events among taxa. Woodruff (2003) identified the need for phylogeographic (sensu Avise et al, 1987) or phylogenetic studies of ‘appropriate’ taxa at the Isthmus of Kra interface to further investigate causal mechanisms leading to the biogeographic patterns observed. To date, no such studies have been carried out.
Macrobrachium rosenbergii, the giant freshwater prawn, is an ideal model species for investigating these mechanisms. Molecular analyses of freshwater-dependent taxa should prove particularly useful in this regard, as such organisms are likely to have remained effectively isolated in discrete freshwater drainages after the seaways subsided (unlike amphibians, mammals, birds, etc), limiting their opportunity for range expansion or secondary contact that could make interpretation of the data difficult. M. rosenbergii has a broad distribution in the region, and our previous study (de Bruyn et al, 2004) indicated that stocks found either side of Huxley's extension of Wallace's Line may have been strongly influenced by the historical geography of the region. Here, we utilise intraspecific mitochondrial DNA (mtDNA) variation in the western form (sensu de Bruyn et al, 2004) of the giant freshwater prawn, M. rosenbergii, to test for evidence for ancient seaways that are believed to have dissected the Thai-Malay Peninsula. Furthermore, we explore the utility of this intraspecific approach (ie phylogeography sensu Avise et al, 1987) for testing biogeographic hypotheses.
Methods
Taxa, sample collection and molecular analyses
M. rosenbergii is an obligate freshwater crustacean as an adult but requires brackishwater for larval survival and development. Results of salinity-tolerance experiments on M. rosenbergii suggest that both adults and postlarvae can survive in brackish conditions (up to 12 ppt) for extended periods of time without any apparent detrimental effects. They are unable, however, to tolerate full marine conditions for more than a week as adults and 20 days as postlarvae (Sandifer et al, 1975). Prawns used in this study were collected from localities indicated in Figure 1 and Table 1, and were identified using Short's Macrobrachium key (1998). As M. rosenbergii are a commercially important species (FAO, 2000), a factor that constrained selection of sampling sites for this study (particularly for sites adjacent to the postulated seaways) was the need for sites to be free of translocated stock to eliminate the potential that prawns with non-native genotypes were included in the analysis (Thai Freshwater Fisheries; N Pongthana, pers. comm.). This resulted in an unbalanced sampling design either side of the postulated seaways, that is, more ‘southern’ than ‘northern’ sites. This lack of balance does not diminish the findings of this study, as it has been demonstrated that sampling of at least 90 individuals (from uncontaminated wild stocks collected from either side of the proposed seaways) provides the statistical power to detect with 95% probability at least one copy of all haplotypes occurring at a frequency of 1% (Schwager et al, 1993).
Tissue samples (muscle or pleopod) were stored in 70% ethanol until required for molecular analyses. For DNA extraction, a small piece of tissue was first rehydrated for 30 min in 1 ml GTE buffer (100 mM glycine, 10 mM Tris, 1 mM EDTA). Tissue samples were then incubated overnight at 55°C in 500 μl extraction buffer (100 mM NaCL, 50 mM Tris, 10 mM EDTA, 0.5% SDS) containing 20 μl of 10 μg/μl Proteinase K (Sigma Co.). Total genomic DNA was extracted using standard phenol : chloroform extraction methods, and collected by ethanol precipitation. Amplification of a fragment of the mtDNA cytochrome c oxidase subunit I (COI) gene was carried out using primers LCO1490 and HCO2198 (Folmer et al, 1994). PCR conditions were as follows: each 50 μl amplification reaction consisted of 400 ng of template DNA, 5 μl of 10 × buffer containing MgCl2 (Roche), an additional 2 μl of 25 mM MgCl2 (Roche), 0.5 units of Taq polymerase (Roche), 0.8 μl of each primer (10 μM final conc.), 0.2 mM of each dNTP, and 38.95 μl autoclaved ddH2O. Samples that proved difficult to PCR were amplified using READY-TO-GO®BEADS (Pharmacia Biotech). Thermal cycling was performed on a PTC-100 thermocycler (MJ Research Inc.) under the following conditions: 3 min denaturation at 94°C, followed by 30 cycles of 30 s at 94°C, 30 s at 55°C, 30 s at 72°C, and a final 10 min extension at 72°C, before cooling to 4°C for 10 min. Negative controls were included in all PCR runs. PCR amplifications were confirmed with agarose gel electrophoresis on a 1% gel. Screening for intrapopulation variation was carried out using Temperature Gradient Gel Electrophoresis (TGGE) combined with Outgroup Heteroduplex Analysis (OHA) (Campbell et al, 1995). This method proved to be sensitive enough to consistently distinguish among haplotypes that varied by a single base pair (bp). All individuals were analysed by way of TGGE/OHA, and two to three individuals exhibiting identical banding patterns from each population were sequenced to confirm that they shared identical haplotypes. PCR products from haplotypes identified as unique using TGGE/OHA were purified using a Qiagen QIAquick PCR purification kit. DNA sequencing of 602 bp of the COI gene was conducted on an ABI 3730 automated sequencer at the Australian Genome Research Facility at the University of Queensland, Brisbane, Australia. Both strands of the PCR product were completely sequenced.
Data analysis
Sequences were aligned in ClustalX (Thompson et al, 1997) with parameters set to default. Initial data exploration and Kimura 2-parameter (Kimura, 1980) sequence divergences were carried out in MEGA ver. 2.1 (Kumar et al, 2001). Haplotype (h) and nucleotide diversity (π) indices and Tajima's D test (Tajima, 1989) for neutrality were performed in DnaSP (Rozas et al, 2003). A bootstrapped (1000 pseudoreplicates; Felsenstein, 1988) neighbour-joining (Saitou and Nei, 1987) phylogenetic tree was estimated in MEGA to identify levels of statistical support for discrete clades identified. An M. rosenbergii individual sampled from Bali was identified in an unpublished (de Bruyn, unpublished data) 16S mtDNA phylogeny as an appropriate outgroup to root the tree. To test for adherence to a clock-like evolution of the mtDNA sequences, a log-likelihood ratio test was carried out in PAUP* 4.0b10 (Swofford, 2002) that compared trees generated under the assumption of a molecular clock, to trees unconstrained by any such assumption (Felsenstein, 1988). The timing of cladogenesis identified in the phylogeny was then inferred by way of molecular clock approximation.
Geographical associations among haplotypes were tested using nested clade analysis (NCA; Templeton, 1998). A haplotype cladogram was generated in TCS (Clement et al, 2000), and then manually converted into a nested design using the nesting rules outlined in Templeton and Sing (1993), Crandall (1996) and Templeton (1998). This nested design was then analysed in GeoDis ver. 2.0 (Posada et al, 2000) with the null hypothesis of no geographic association among haplotypes. We made a qualitative decision to use geographic coordinates to determine distances among sites, as opposed to stream distance (Fetzner and Crandall, 2003), as most sampling sites were restricted to geographically isolated drainage basins. Templeton's (2004) latest inference key was used to infer processes involved in any statistically significant associations observed. It has recently been suggested that some NCA inferences may be flawed, and should therefore be supported by the use of alternative analytical techniques (eg Alexandrino et al, 2002; Masta et al, 2003; Templeton, 2004). We therefore applied the coalescent/maximum-likelihood approach implemented in FLUCTUATE ver. 1.4 (Kuhner et al, 1998) to determine if there was evidence for population expansion events in clades identified with NCA. Specifically, the exponential growth or decline of the population can be inferred by positive or negative values of the exponential growth parameter g. An exploratory search strategy implementing 20 short chains of 1000 steps each, and five long chains of 20 000 steps were used to determine parameters for the production runs. Production runs were implemented using 20 short chains of 8000 steps each, and 10 long chains of 50 000 steps. The program was run multiple times to ensure concordance of parameter estimates.
Results
In total, 404 M. rosenbergii individuals (excluding the outgroup) were analysed for variation in a 602 bp fragment of the mtDNA COI gene using TGGE/OHA analyses. Representatives (2–3) from each population that displayed identical banding patterns were sequenced to confirm they shared identical haplotypes, which was confirmed in all cases. This resulted in the identification of 35 putative haplotypes (GenBank Accession numbers: AY554293-AY554327), defined by 54 segregating sites. No significant deviations from neutrality were identified in our dataset (Tajima's D=−1.284, P>0.10). Nucleotide substitutions favoured transitions over transversions, yielding a transition/transversion ratio of 4.16. The neighbour-joining phylogeny strongly supported the existence of two widely distributed monophyletic clades situated approximately 120 km apart on the Isthmus of Kra (bootstrap values: 91% for southern clade, 94% for northern clade). Similar support was observed in the 95% probability cladogram (Figures 1 and 2), with populations sampled from sites north and south of the more northerly seaway restricted to two distinct monophyletic clades, except for site c (Kraburi River, SW Thailand) situated just north of the northern seaway, which is characterised by individuals exhibiting both ‘northern’ and ‘southern’ haplotypes (Figure 1 and Table 2). NCA identified an allopatric fragmentation event followed by range expansion at the highest nesting level, that is, between northern and southern clades either side of the hypothesised northern seaway (Table 3). Subsequently, we performed Templeton's supplementary test for secondary contact (Templeton, 2001), which confirmed this hypothesis.

95% probability cladogram estimated from the M. rosenbergii COI data. Small black circles indicate inferred missing haplotypes not observed in the dataset. Haplotype 29 was the haplotype with the highest frequency (n=134) and the highest root probability (Castelloe and Templeton, 1994). See Figure 1 and Table 2 for haplotype frequencies.
Within the northern clade, NCA suggested contiguous range expansion at both ancestral and younger clade levels, that is, clades 3–2 and 1–5, respectively, and restricted gene flow with isolation-by-distance at the 1-step level for clades 1–3 (Table 3). Within the southern clade, restricted gene flow with some long distance dispersal was suggested at both ancestral and younger clade levels, that is, clades 2–7 and 1–9 respectively, while at the 1-step level contiguous range expansion (clades 1–14) and restricted gene flow with isolation-by-distance (clades 1–15) were also inferred (Table 3).
FLUCTUATE analyses of maximum likelihood estimates of g supported the NCA inferences of expansion events in clades 3–2 (g=235.1±2 s.d. 67.3) and 1–5 (g=303.3±2 s.d. 64.0). Similarly, concordant patterns in FLUCTUATE and NCA suggested no evidence for growth in clades 2–7 (g=−26.1±2 s.d. 45.6) and 1–9 (g=−160.9±2 s.d. 76.2). Accurate testing of clades 1–3, 1–14 and 1–15 were precluded by the star-like structure of these clades. Such patterns result in equally high values of Θ and g, which cause the program to fluctuate wildly on the likelihood surface until estimates become huge and the program ‘overflows’ (Kuhner, 2003). Implementing the analyses with more steps in each chain, and more chains (Kuhner, 2003) did not alter this outcome.
A fairly low level of divergence was evident within clades (Table 2; Figure 2), particularly taking into account the considerable geographic distances among sites, for example, Bangladesh to SW Thailand (∼700 km). Kimura 2-parameter sequence divergences ranged from 0.002 to 0.015 within clades to 0.019–0.031 between the two clades. Genetic diversity measures were similar in the two clades: northern clade (h=0.49, π=0.00619), southern clade (h=0.51, π=0.00593). A log-likelihood ratio test could not reject the hypothesis that lineages were evolving according to a clock-like model of evolution (−ln L=1261.43 with molecular clock enforced vs −ln L=1247.17 without molecular clock enforced, χ2=47.4, d.f.=33, P>0.10). A COI molecular clock rate of 1.4%/Myr (Knowlton and Weigt, 1998) based on the smallest sequence divergence observed among 15 pairs of ‘geminate’ snapping shrimp taxa, presumably separated by the closure of the Isthmus of Panama seaway, is commonly used as a calibration point for estimating divergence times in phylogeographic studies. Recent studies, however, warn against taking these estimates at face-value, due to a number of factors known to bias such estimates (see Marko (2002) and references therein for discussion). Indeed, Knowlton and Weigt's (1993) earlier COI molecular clock estimates were found to be erroneous (Knowlton and Weigt, 1998). A further complication is the recent finding that some so-called ‘geminate’ pairs may not be each other's closest living relatives, and their divergence may in fact not be related to the closure of the Panamanian seaway (Craig et al, 2004). It has therefore been suggested that calibrations based on the fossil record may be more appropriate, although the lack of a suitable fossil record for shrimp/prawns make this approach problematic. Nonetheless, we approximated a rough estimate of divergence time between the northern and southern clades based on Knowlton and Weigt's (1998) mtDNA COI molecular clock (1.4%/Myr), and a single crustacean mtDNA COI calibration based on the fossil record (0.13–0.55%/Myr; Schön et al, 1998) in an attempt to minimize error in our inferences. The geological calibration suggested a minimum divergence time in the region of 2.2–1.3 Mya, while the fossil record calibration provides a much more conservative estimate of 14.1–3.3 Mya.
Discussion
If dispersal (gene flow) of freshwater taxa has been restricted in the past by a significant geographic barrier that lead to cladogenesis (vicariance) among populations situated north and south of the proposed seaways, a molecular signature of allopatric fragmentation between these populations would be expected. Moreover, if contemporary gene flow has resulted in secondary contact between northern and southern populations since subsidence of the marine barriers, this should be evident in two monophyletic clades that overlap in their geographic distribution, indicating patterns of secondary intergradation. Results presented here indicate a sharp genetic break between M. rosenbergii populations situated on the Isthmus of Kra. Our analyses support the hypothesis that an ancient marine seaway divided the Thai-Malay Peninsula, resulting in a vicariant event that restricted gene flow among populations either side of this divide. All populations (except site c) north and south of the northern proposed seaway, separated by a distance of only 120 km, belong to two widely distributed monophyletic mtDNA clades that were apparently restricted to either side of this seaway (Figures 1 and 2). If the genetic break evident in our data is indeed a result of the Pliocene-era seaway (5.5–4.5 Mya; Woodruff, 2003), and not a later unidentified rise in sea-level, the results of our molecular clock analyses would suggest that either: (i) rates of COI evolution in M. rosenbergii are significantly slower than in snapping shrimp, or (ii) the mtDNA COI clock rate calibrated to the closure of the Panamanian seaway (Knowlton and Weigt, 1998) is too great. No matter the case, our results suggest that the use of such calibrations should be applied cautiously when a good predictive temporal framework (eg used in this study) is not available.
The Kraburi River population (site c) just north of the seaway was unique because it possessed both ‘northern’ haplotypes and a single ‘southern’ haplotype, indicating a recent northward expansion of the southern clade into this northern site leading to admixture of the two groups (Figure 1). This molecular signal of a recent range expansion event into the Kraburi River (site c) is indicated by the occurrence of nine ‘northern’ haplotypes found in low frequencies (four are singletons; see Table 2), and the presence of a single southern haplotype at a high frequency (n=26; haplotype 21), suggesting a recent founder event followed by local self-seeding (Mayr, 1942). This scenario was strongly supported by the NCA (Table 3). There is no explicit support in our data for the second and more southerly seaway, however this may be a consequence of the lesser width of this seaway (Figure 1) and the dispersal capabilities of the organism in question.
Because populations that are today geographically isolated from each other by a marine environment share haplotypes over distances of hundreds of kilometres, the question arises as to what mechanisms are involved in maintaining these relationships. In a previous phylogenetic study conducted at a broader spatial scale (de Bruyn et al, 2004), it was suggested that Pleistocene drainage basins that linked sites on the Sunda Shelf that are today geographically isolated, may have acted as conduits for gene-flow among some populations of M. rosenbergii. Similar studies on freshwater fish indicate the important role that ancient drainage basins have played in shaping the distribution of molecular variation in freshwater organisms (Hurwood and Hughes, 1998; Waters et al, 2001; Kotlik et al, 2004 and others). This ancient drainage basin hypothesis goes some way to describing the close relationship among populations observed here, particularly for sites d–h (ie southern Thai-Malay Peninsula and Sth. Vietnam). These sites are likely to have been linked by freshwater via the Siam or Malacca Straits River Systems that existed during the Pleistocene (Voris, 2000; see de Bruyn et al, 2004 for discussion). NCA provides support for this scenario, inferring a fairly recent (in evolutionary terms; 1-step clade level) contiguous range expansion among all southern clade sites except for SE Kalimantan. The Kalimantan population (site k) appears to have been isolated historically for an extended period of time, as there is virtually no sharing of haplotypes with any other southern sites (Table 2). Interestingly, SE Kalimantan is the population involved in both recent and ancestral inferred events utilising NCA; restricted gene flow with some long distance dispersal, and restricted gene flow with isolation-by-distance. This pattern warrants further investigation.
The close genetic relationship between the Sumatran (site j) and Mekong River (site i) populations, and all other populations in the southern clade, however, cannot be fully explained by Pleistocene drainage basins. Along similar lines, the close genetic relationship between northern clade populations from Bangladesh (sites a & b) and Thailand (site c) cannot be explained by inferring past gene-flow via freshwater systems, as these geographically distant sites have no history of a freshwater connection. Freshwater plumes from the extensive Ganges system may explain gene flow between SE and SW Bangladesh, but is unlikely to explain long distance gene flow between Bangladesh and Thailand (Dai and Trenberth, 2002), some 700 km distant. NCA again identified biologically feasible processes that may have resulted in the population genetic structuring observed in the northern clade, that is, historical contiguous range expansion between SW Thailand and Bangladesh, and recent contiguous range expansion accompanied by restricted gene flow with isolation-by-distance among all northern sites. At this stage, this hypothesis must remain mere conjecture (while supported by the NCA) as no field work could be undertaken in Myanmar so the isolation-by-distance effect observed in the northern clade may simply result from inadequate sampling in this region.
Previous work on freshwater organisms (Waters and Burridge, 1999; Waters et al, 2000; McDowall, 2002) has highlighted the largely unrecognised role marine dispersal can play in the evolution of some ‘freshwater’ aquatic taxa. As M. rosenbergii are estuarine dependent, and all life-stages tolerate full marine conditions to varying degrees, a stepping-stone model of gene flow (Kimura and Weiss, 1964) via the marine environment between adjacent estuaries, accompanied by occasional long-distance marine dispersal during favourable conditions may best explain the observed population structure of this species. To clarify the role of marine dispersal, drainage basin maps of Thailand were examined to identify potential routes for southern to northern clade colonisation, as observed at the Kraburi River site (site c). No indication of a possible freshwater colonisation route between southern and northern sites was found, suggesting a marine dispersal route. In addition, if dispersal was via freshwater, and not a ‘rare’ marine dispersal event, we might expect to find evidence for bi-directional movement of haplotypes, that is, south-north and vice versa, or at the very least presence of more than a single southern haplotype at the Kraburi River site. Instead, we observed evidence for unidirectional (south to north) movement, and fixation of a single derived (tip) southern haplotype at this site. A future analysis of populations sampled from a number of adjacent estuaries will be required to determine the role of marine dispersal in this species.
The results presented here provide the first molecular support for the existence of an ancient biogeographic barrier, the Isthmus of Kra Seaway. Molecular evidence indicates that this seaway was extensive enough to restrict gene flow in M. rosenbergii, a ‘freshwater’ crustacean that may be capable of some marine dispersal. Since the time when the seaway subsided, contemporary gene flow appears to have occurred across this historical barrier, highlighting the mutually compatible roles that both vicariance and dispersal have played in the evolutionary history of M. rosenbergii. Our results emphasise the importance of choosing an appropriate model organism, and the power provided by a phylogeographic approach, in testing historical biogeographical hypotheses.
References
Alexandrino J, Arntzen JW, Ferrand N (2002). Nested clade analysis and the genetic evidence for population expansion in the phylogeography of the golden-striped salamander, Chioglossa lusitanica (Amphibia: Urodela). Heredity 88: 66–74.
Avise JC, Arnold J, Ball RM, Bermingham E, Lamb T, Neigel JE et al (1987). Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Ann Rev Ecol Syst 18: 489–522.
Bermingham E, McCafferty SS, Martin A, Kocher TD, Stepien CA (1997) (eds) Fish biogeography and molecular clocks: perspectives from the Panamanian Isthmus. In Molecular Systematics of Fishes. Academic Press: San Diego, London, pp 113–128.
Campbell NJH, Harriss FC, Elphinstone MS, Baverstock PR (1995). Outgroup heteroduplex analysis using temperature gradient gel electrophoresis: high resolution, large scale, screening of DNA variation in the mitochondrial control region. Mol Ecol 7: 407–418.
Castelloe J, Templeton AR (1994). Root probabilities for intraspecific gene trees under neutral coalescent theory. Mol Phyl Evol 3: 102–113.
Clement M, Posada D, Crandall KA (2000). TCS: a computer program to estimate gene genealogies. Mol Ecol 9: 1657–1659.
Collins LS, Budd AF, Coates AG (1996). Earliest evolution associated with closure of the Tropical American Seaway. Proc Natl Acad Sci USA 93: 6069–6072.
Corbet AS (1941). The distribution of butterflies in the Malay Peninsula. Proc R Entom Soc Lond A 16: 101–116.
Corbett GB, Hill JE (1992). The Mammals of the Indomalayan Region: a Systematic Review. Oxford University Press: Oxford, UK.
Craig MT, Hastings PA, Pondella DJ (2004). Speciation in the Central American Seaway: the importance of taxon sampling in the identification of geminate species pairs. J Biogeog 31: 1085–1091.
Crandall KA (1996). Multiple interspecies transmissions of human and simian T-cell leukemia/lymphoma virus type I sequences. Mol Biol Evol 13: 115–131.
Dai A, Trenberth KE (2002). Estimates of freshwater discharge from continents: latitudinal and seasonal variations. Jnl Hydrometeorol 3: 660–687.
de Bruyn M, Wilson JC, Mather PB (2004). Huxley's Line demarcates extensive genetic divergence between eastern and western forms of the giant freshwater prawn, Macrobrachium rosenbergii. Mol Phyl Evol 30: 251–257.
Denduangboripant J, Cronk QCB (2000). High intraindividual variation in internal transcribed spacer sequences in Aeschynanthus (Gesneriaceae): implications for phylogenetics. Proc R Soc Lond B 267: 1407–1415.
FAO (Food Agriculture Organisation of the United Nations (2000). Aquaculture production statistics 1989–1998. FAO Fisheries Circular 815 (Rev 12). FAO: Rome.
Felsenstein J (1988). Phylogenies from molecular sequences: inference and reliability. Ann Rev Genet 22: 521–565.
Fetzner Jr JW, Crandall KA (2003). Linear habitats and the nested clade analysis: an empirical evaluation of geographic versus river distances using an Ozark crayfish (Decapoda: Cambaridae). Evolution 57: 2101–2118.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 3: 294–299.
Hanebuth T, Stattegger K, Grootes PM (2000). Rapid flooding of the Sunda Shelf: a late-glacial sea-level record. Science 288: 1033–1035.
Hoffman FG, Baker RJ (2003). Comparative phylogeography of short-tailed bats (Carollia: Phyllostomidae). Mol Ecol 12: 3403–3414.
Hughes JB, Round PD, Woodruff DS (2003). The Indochinese-Sundaic faunal transition at the Isthmus of Kra: an analysis of resident forest bird species distributions. J Biogeog 30: 569–580.
Hurwood DA, Hughes JM (1998). Phylogeography of the freshwater fish, Mogurnda adspersa, in streams of northeastern Queensland, Australia: evidence for altered drainage patterns. Mol Ecol 7: 1507–1517.
Inger RF (1966). The systematics and zoogeography of the amphibia of Borneo. Field Zool 52: 1–402.
Inger RF, Voris HK (2001). The biogeographical relations of the frogs and snakes of Sundaland. J Biogeog 28: 863–891.
Kimura M (1980). A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. Jnl Mol Evol 16: 111–120.
Kimura M, Weiss GH (1964). The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49: 561–576.
Knowlton N, Weigt LA, Solorzano LA, Mills DK, Bermingham E (1993). Divergence in proteins, mitochondrial DNA, and reproductive compatibility across the Isthmus of Panama. Science 260: 1629–1632.
Knowlton N, Weigt LA (1998). New dates and new rates for divergence across the Isthmus of Panama. Proc R Soc Lond B 265: 2257–2263.
Kotlik P, Bogutskaya NG, Ekmekci FG (2004). Circum Black Sea phylogeography of Barbus freshwater fishes: divergence in the Pontic glacial refugium. Mol Ecol 13: 87–96.
Kuhner MK, Yamato J, Felsenstein J (1998). Maximum likelihood estimation of population growth rates based on the coalescent. Genetics 149: 429–434.
Kuhner MK (2003). LAMARC: estimating population genetic parameters from molecular data. In: Salemi M, Vandamme A (eds) The Phylogenetic Handbook: a Practical Approach to DNA and Protein Phylogeny. Cambridge University Press: Cambridge, pp 379–399.
Kumar S, Tamura K, Jakobsen IB, Nei M (2001). MEGA2: Molecular Evolutionary Genetics Analysis Software. Arizona State University: Tempe, AZ, USA.
Marko PB (2002). Fossil calibration of molecular clocks and the divergence times of geminate species pairs separated by the Isthmus of Panama. Mol Biol Evol 19: 2005–2021.
Masta SE, Laurent NM, Routman EJ (2003). Population genetic structure of the toad Bufo woodhousii: an empirical assessment of the effects of haplotype extinction on nested cladistic analysis. Mol Ecol 12: 1541–1554.
Mayr E (1942). Systematics and the Origin of Species. Columbia University Press: New York, USA.
McDowall RM (2002). Accumulating evidence for a dispersal biogeography of southern cool temperate freshwater fishes. J Biogeog 29: 207–219.
Posada D, Crandall KA, Templeton AR (2000). GeoDis: a program for the cladistic nested analysis of the geographical distribution of genetic haplotypes. Mol Ecol 9: 487–488.
Ridder-Numan JWA (1998). Historical biogeography of Spatholobus (Leguminosae-Papillionoideae) and allies in SE Asia. In: Hall R, Holloway JD (eds) Biogeography and Geological Evolution of Southeast Asia. Backhuys: Leiden, Netherlands, pp 259–277.
Rozas J, Sanchez-DelBarrio JC, Messueger X, Rozas R (2003). DnaSp, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497.
Saitou N, Nei M (1987). The neighbour-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–426.
Sandifer PA, Hopkins JS, Smith TIJ (1975). Observations on salinity tolerance and osmoregulation in laboratory-reared Macrobrachium rosenbergii post-larvae (Crustacea: Caridea). Aquaculture 6: 103–114.
Schön I, Butlin RK, Griffiths HI, Martens K (1998). Slow molecular evolution in an ancient asexual ostracod. Proc R Soc Lond B 265: 235–242.
Schwager SJ, Mutschler MA, Federer WT, Scully BT (1993). The effect of linkage on sample size determination for multiple trait selection. Theor Appl Genet 86: 964–974.
Short J (1998). Pictorial Key to Australian Macrobrachium. Queensland Museum Publication, Brisbane, Australia.
Swofford DL (2002). PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4. Sinauer Associates: Sunderland, MA.
Tajima F (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Templeton AR (1998). Nested clade analyses of phylogeographic data: testing hypotheses about gene flow and population history. Mol Ecol 7: 381–397.
Templeton AR (2001). Using phylogeographic analyses of gene trees to test species status and processes. Mol Ecol 10: 779–791.
Templeton AR (2004). Statistical phylogeography: methods of evaluating and minimizing inference errors. Mol Ecol 13: 789–810.
Templeton AR, Sing CF (1993). A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. IV. Nested analyses with cladogram uncertainty and recombination. Genetics 134: 659–669.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgens DG (1997). The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 24: 4876–4882.
Tringali MD, Bert TM, Seyoum S, Bermingham E, Bartolacci D (1999). Molecular phylogenetics and ecological diversification of the transisthmian fish genus Centropomus (Perciformes: Centropomidae). Mol Phyl Evol 13: 193–207.
Vail PR, Hardenbol J (1979). Sea-level changes during the tertiary. Oceanus 22: 71–79.
Voris HK (2000). Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. J Biogeog 27: 1153–1167.
Waters JM, Burridge CP (1999). Extreme intraspecific mitochondrial DNA sequence divergence in Galaxias maculatus (Osteichthys: Galaxidae), one of the world's most widespread freshwater fish. Mol Phyl Evol 11: 1–12.
Waters JM, Dijkstra LH, Wallis GP (2000). Biogeography of a southern hemisphere freshwater fish: how important is marine dispersal? Mol Ecol 9: 1815–1821.
Waters JM, Craw D, Youngson JH, Wallis GP (2001). Genes meet geology: fish phylogeographic pattern reflects ancient, rather than modern, drainage connections. Evolution 55: 1844–1851.
Woodruff DS (2003). Neogene marine transgressions, palaeogeography and biogeographic transitions on the Thai-Malay Peninsula. J Biogeog 30: 551–567.
Zeh A, Zeh DW, Bonilla MM (2003). Phylogeography of the harlequin beetle-riding pseudoscorpion and the rise of the Isthmus of Panama. Mol Ecol 12: 2759–2769.
Acknowledgements
We thank Kriket Broadhurst, Daisy Wowor, Peter Ng, Nuanmanee Pongthana and colleagues, Nguyen Van Hao, Tran Ngoc Hai, Pek Yee Tang, Selvaraj Oyyan and Abol Munafi Ambok Bolong for their help in acquiring specimens for this study. Jane Hughes, David Hurwood, Andrew Baker and the EGGers provided helpful discussions on the manuscript. MdB acknowledges support from an Australian Postgraduate Award, and research grants from the Australian Geographic Society, the Ecological Society of Australia and the Linnean Society of New South Wales. PBM acknowledges support from an ACIAR small-project Grant FIS/2002/083 and assistance from regional fisheries agencies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Bruyn, M., Nugroho, E., Hossain, M. et al. Phylogeographic evidence for the existence of an ancient biogeographic barrier: the Isthmus of Kra Seaway. Heredity 94, 370–378 (2005). https://doi.org/10.1038/sj.hdy.6800613
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800613
Keywords
This article is cited by
-
Linking genetic lineages of giant freshwater prawn Macrobrachium rosenbergii to their global populations
Fisheries Science (2024)
-
New freshwater mussel taxa discoveries clarify biogeographic division of Southeast Asia
Scientific Reports (2020)
-
Exploring community assembly among Javanese and Balinese freshwater shrimps (Atyidae, Palaemonidae) through DNA barcodes
Hydrobiologia (2020)
-
Genetic variation, demographic history and phylogeography of tire track eel, Mastacembelus favus (Synbranchiformes: Mastacembelidae) in Southeast Asia
Hydrobiologia (2019)
-
Population genetic structure of the mantis shrimp Oratosquilla oratoria (Crustacea: Squillidae) in the Yellow Sea and East China Sea
Journal of Oceanology and Limnology (2018)
