
Abstract
Using fast-evolving microsatellites, more slowly evolving ITS markers and performing habitat analyses, we demonstrated a drastic genetic divergence and significant habitat differentiation between early- and late-flowering variants of plants morphologically belonging to Gymnadenia conopsea ssp conopsea. The two phenological variants can either be found in separate or in mixed populations. Information from microsatellite markers and ITS sequences indicated the occurrence of an early historical split between the two flowering-time variants, a split that has been maintained until the present time even within sympatric populations. Early-flowering variants were also far more genetically diverse, had more alleles per microsatellite locus and a wider habitat amplitude than late-flowering variants. As a comparison, we included G. odoratissima in the sequencing study. We found G. odoratissima to be most closely related to the early-flowering type. This indicates a more ancient divergence event between the two flowering-time variants within G. conopsea ssp conopsea than between the two different species G. odoratissima and the early-flowering variant of G. conopsea. Possible explanations to the results arrived at and possible mechanisms maintaining the genetic separation are discussed.
Similar content being viewed by others


Introduction
The level of gene flow between populations determines to what extent individuals within a species share a common gene pool as well as the amount of local genetic subdivision (Mayr, 1970). Gene flow is often seen as a constraining force in evolution (Mayr, 1970; Slatkin, 1987). In natural populations, selection acts in adapting populations to conditions in the local environment. Therefore, gene flow in the form of immigrants from other populations with genes adapted to other conditions could counteract the ongoing selection process (Slatkin, 1987; Lenormand, 2002). On the other hand, with an interrupted or restricted gene flow, populations could, both by chance and as a result of different environmental conditions, evolve independently and as a consequence eventually become incompatible with each other, which could result in the creation of new species (eg Mayr, 1970; Turelli et al, 2001).
The understanding of plant speciation processes has greatly increased during the last 50 years (see Levin, 2001). However, a fundamental problem is determining the relative importance of the many complex mechanisms involved in the formation of new species (Rundle and Whitlock, 2001). One way of approaching the problem could be focusing on maintenance of species identity, that is, to identify the biological barriers or reproductive isolation mechanisms that keep species discrete in sympatry (Rundle and Whitlock, 2001). Slatkin (1987) discusses three components appearing in the formation of new species of which restrictions in gene flow is one. The other two are evolution of reproductive isolating mechanisms and accumulation of morphological and behavioural differences.
One example of a possible isolating mechanism in plants is differentiation in flowering time. The strength of the barrier depends on the degree of separation. In some species or populations it appears as a prolonged flowering period during which individuals do overlap in flowering time, whereas in others distinct early and late phenological variants are observed. The former may result in decreased genetic exchange between variants, but it will probably not have crucial effects on genetic structure; however, strong temporal isolation, genetic drift and/or selection, could enhance genetic divergence and further strengthen differentiation between the flowering-time variants, ultimately resulting in the evolution of new species. Differentiation in flowering time has been observed within several plant taxa, for example, in the genera Euphrasia (Karlsson, 1984), Beta (Van Dijk et al, 1997), Gentianella (Lennartsson, 1997), Silene (Hauser and Weidema, 2000), Capsella (Neuffer and Hurka, 1999); see Lennartsson (1997) for additional taxa.
In the orchid family, variation in flowering phenologies has been described in Gymnadenia conopsea, the fragrant orchid (Heusser, 1938). Besides flowering phenology, this species is highly variable also in many other characters (Soliva and Widmer, 1999). The species is, in Sweden, divided into two phenologically and morphologically distinguishable varieties or subspecies, var/ssp conopsea and var/ssp densiflora (Mossberg and Stenbeck, 1992; Krok and Almquist, 1994). The late-flowering densiflora flowers (in southern Sweden) around the middle of July, whereas conopsea is early-flowering (in southern Sweden around mid-June).
In several populations, individuals that are morphologically alike the early-flowering variety but still late-flowering have been found to co-flower with the typical densiflora variety. In areas where all three types can be found, populations can either be exclusively early- or late-flowering or mixed, with individuals with different flowering times growing side by side. We wanted to know if those types are separated genetically and if they have different habitat preferences.
To discriminate between both recent and long-term events, we selected genetic markers with contrasting mutation rates, fast-evolving microsatellite loci, and the internal transcribed spacer (ITS) region sequences with a much lower mutation rate. For comparison with closely related taxa, some individuals of G. conopsea ssp densiflora and some individuals of the rare congener Gymnadenia odoratissima were included in the ITS study.
From three Swedish regions, one mainland and the major Baltic islands of Öland and Gotland, we used 17 populations to test whether flowering-time types have different habitat preferences and if they are differentiated within each population or belong to historically different evolutionary lineages, within regions or within Sweden that are kept distinct in sympatry.
Materials and methods
The study species
G. conopsea ssp conopsea, the fragrant orchid, is a perennial, terrestrial orchid. It is generally found in calcareous areas, like open grasslands, grazed meadows and close to marshes and fens. The height is about 10–40 cm (Mossberg and Stenbeck, 1992). Subspecies densiflora prefers more moist habitats and it usually grows close to calcareous chars (Mossberg and Stenbeck, 1992). This variant is taller (up to 70 cm), has broader leaves (ca 2 cm) and is more densely flowered compared to conopsea (Mossberg and Stenbeck, 1992). In both types, the flower colour varies from pale pink to cerise or lilac and rarely to pure white. The flowers are heavily scented, and pollinators, different species of Lepidoptera, are rewarded with an abundant amount of nectar (Proctor et al, 1996). The geographic distribution covers most of Europe and parts of Asia. In Sweden, the species is quite rare but it has a wide distribution, from Scania in the south to Torne Lappmark in the north (densiflora is only found in southern Sweden).
Populations
Plant material was collected from 17 Swedish populations of G. conopsea ssp conopsea (Figure 1): seven populations from the island of Gotland, seven from the island of Öland, and three from the province of Västergötland. Seven localities were either early-flowering (Öjs mosse, Svenshultsby and Svartarpskärret) or late-flowering type (Hoburgsmyr, Brucebo, Grausne and Torpmossen), whereas 10 localities (Lojsta, Besteträsk, Horsan, Klinte, Grossemyr, Persnäs, Gråborg, Amunds mosse, Sandby and Skogatorpskärret) were mixed populations with both flowering-time variants. Individuals of G. conopsea ssp densiflora were sampled from four populations (Amundsmosse, Gråborg, Sandby and Skogatorpskärret). For additional information, one individual from six different populations of G. odoratissima (four populations from the island of Gotland and one from each of the provinces of Västergötland and Östergötland) was included in the ITS sequencing study.

Locations of G. conopsea populations included in the study.
DNA extraction
In the sampling procedure, a small piece of leaf was collected and stored in silica gel at room temperature. The DNeasy Plant Mini Kit (Qiagen) was used for DNA extractions (according to the manufacturer's recommendations).
Microsatellites
A total of 3–10 individuals of each flowering-time type and locality were analysed at four microsatellite loci (Gc29, Gc31, Gc42 and Gc51), developed for the fragrant orchid Gustafsson and Thorén (2000), Gc31 not included in the referred article), giving a total of 27 sampled populations from 17 localities.
One of the two primers in each locus was end-labelled using T4 polynucleotide kinase (Promega) and γ33P ATP (Amersham) in a reaction volume of 10 μl (enough for 90 reactions) consisting of 1 × buffer (Promega), 5 U T4 polynucleotide kinase (Promega), 20 pmol primer and 50 μCi γ33P ATP. End-labelling reaction mix was incubated for 30 min at 37oC, followed by 2 min at 90oC (to stop the reaction) using an MJ Research PTC-100 thermal cycler. The alleles were PCR amplified in 10 μl reaction volumes. The reaction mixture consisted of 1 μl template DNA solution, 1 μl 10 × Taq buffer (MBI Fermentas), 0.4 U Taq DNA polymerase (MBI Fermentas), 1.2–1.5 mM MgCl2, 4 pmol nonradioactive primer, 2.5 nmol of each dNTP, 20 μg Spermidine or BSA, 0.22 pmol end-labelled primer +3.78 pmol of the same primer that was not end-labelled. PCR conditions were one denaturing step at 94°C for 3 min followed by 30–35 cycles consisting of 94°C for 30 s, Tao for 30 s and 72°C for 45 s (Tao=62, 60, 52 and 60°C at loci Gc29, Gc31, Gc42 and Gc51, respectively). The cycles were followed by a final elongation step at 72°C for 10 min. The PCR product was mixed with 7 μl formamide and loading dye, denatured in 90°C for 5 min and loaded in a 6% polyacrylamide (PAA) sequencing gel (8 M urea).
Sequencing
One individual (in Klinte two individuals) of each flowering type and population, six individuals from four populations of densiflora and one individual from each G. odoratissima population were chosen for ITS sequence analysis.
The DNA region including 5.8S rDNA and the transcribed spacers on either side, ITS 1 and 2, were PCR amplified using an MJ Research PTC-100 thermal cycler. Primers a and d of Leskinen and Pamilo (1997) were used for PCR amplification. PCR was performed in 50 μl reaction volumes containing 5 μl template DNA solution, 1 X Taq buffer (MBI Fermentas), 2.5 U Taq DNA Polymerase (MBI Fermentas), 1.2 mM MgCl2, 0.2 mM dNTP, 20 ng/μl BSA and 0.25 μmol of each primer. Two PCR reactions were performed for each sample. PCR conditions were one denaturing step at 94°C for 5 min, followed by 35 cycles consisting of denaturing in 94°C for 1 min 10 s, annealing at 54°C for 50 s and elongation at 72°C for 1 min 30 s. The cycles were followed by a final elongation step at 72°C for 10 min. The two PCR products were pooled to increase the final concentration and purified using columns (Qiaquick PCR purification kit, Qiagen). PCR products were sequenced using a ‘Thermo sequenase fluorescent labelled primer cycle sequencing kit’ utilizing 7-deaza-dGTP (Amersham Biosciences). Automated sequencing was performed on an LI-COR DNA Sequencer 4200. Sequences were analysed with the computer program Base ImageIR Ver. 4 and they were aligned manually. The sequences were compared using the program MEGA version 2.1 (Kumar et al, 2001). In calculating pairwise distances, the estimations of the number of nucleotide substitutions per nucleotide site were based on the Kimura two-parameter model. Bootstrap analyses were based on 1000 replications. A UPGMA consensus tree was constructed.
Habitat analyses
Vegetation properties and associated species presence/absence were used to describe niche differentiation between the flowering types (see Prentice and Cramer, 1990). Only localities with mixed types were used in the habitat analyses to avoid confusion with geographical variation in habitat parameters; hence, the analyses will show local, within locality, differentiation in habitat preference. Soil depths were taken and the separate cover percentages of mosses, grasses, Carex and herbs were estimated within a circle of 0.10 m radius around each sampled individual. The presence or absence of Briza media, Eriophorum vaginatum, Filipendula vulgaris, Plantago lanceolata, Potentilla erecta, Sesleria cearulea, Trifolium montanum and T. repens was recorded.
Habitat differentiation between early- and late-flowering individuals was described using a linear discriminant analysis(LDA). The significance of each habitat term was tested in a generalized linear model(GLM) with an assumed binomial error distribution for the response variable flowering time, with the levels early and late. The approach was to create a minimal adequate model (Crawley, 1993). Explanatory variables were first eliminated from the saturated model using the stepAIC procedure (in the MASS library; Venables and Ripley, 1999). This procedure is somewhat more permissive than significance testing by measuring deviance change on omission of the terms one at a time, which was done to further minimize the resulting model. Deviance changes were compared to a χ2 distribution since the dispersion factor (mean residual deviance) was 1.18, which is close to the expected unity. Analyses were made using S-PLUS 2000 (MathSoft, 1999).
Microsatellite data analysis
The observed number of alleles (AO) and expected (HE) heterozygosity and the inbreeding coefficient (FIS) were computed for each population and locus using Genepop 3.1b (Raymond and Rousset, 1995). Expected numbers of heterozygotes were computed using Levene's correction (Li, 1976). FIS values were computed according to the formula of Weir and Cockerham (1984).
Since the number of individuals in the investigated populations were few (3–10 per group), it was not possible to test for linkage disequilibrium and deviations from the Hardy–Weinberg expectations within each population. However, the values were used for comparisons between populations. AO, HE and FIS were used as response variables in linear models where population flowering time was the explanatory variable (assuming Poisson, normal and normal error distribution, respectively, for the three response variables). The frequencies of alleles in each locus were used as explanatory variables in GLMs (binomial error) using populations early- or late-flowering as the response variable. One model was made for each locus and the procedure was as described for the habitat differentiation analysis.
Results
Microsatellite variation
The total numbers of alleles in the four loci and in the two flowering variants were distributed as follows: Gc29–eight alleles in total (eight in the early-flowering populations and six alleles in late-flowering populations); Gc31–four alleles in total (three early, two late); Gc42–19 alleles in total (17 early, two late); Gc51–39 alleles in total (36 early, three late). The GLMs where relative allele frequencies were used to explain the population identity as late- or early-flowering did in three of the loci select one allele that explained virtually all variations in the models. In locus Gc29, allele 5 explained 79.6% of the total deviance. The three alleles that are clearly diagnostic of the late-flowering type are Gc31 allele 4 (explaining 99.9999% of the total deviance), Gc42 allele 2 (99.9997%) and Gc51 allele 19 (99.9999%) (see Figure 2a–d).

Allele distribution among early- and late-flowering variants of G. conopsea ssp conopsea and individuals of a potential ‘hybrid’ origin (cross).
The genetic variability measured as expected heterozygosity (Table 1) was significantly larger in the early-flowering populations for loci Gc31 (F1,23=1.08, P<0.05), Gc42 (F1,23=96, P<0.001) and Gc51 (F1,23=80, P<0.001) but not for locus Gc29 (F1,23=1.08, P=0.3).The number of alleles (Table 2) was significantly higher in early-flowering populations in loci Gc42 (deviance change=30, P<0.001) and Gc51 (deviance change=30, P<0.001). Alleles in loci Gc42 and Gc51 were obviously longer in the early-flowering populations (Figure 2a–d). There were no significant differences in the inbreeding coefficient (FIS) between early- and late-flowering populations. The diagnostic alleles revealed that two of the analysed individuals could be interpreted as ‘hybrids’ between early- and late-flowering types, having one allele each from the two flowering-time variants at all loci, whereas in five other individuals one of four loci expressed an allele from the contrasting flowering-time variant.
Sequence variation
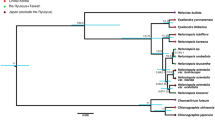
The sequenced regions consisted of 5.8S rDNA and the two flanking spacers ITS 1 and 2.5.8S rDNA is highly conservative and often found to be almost invariant in length in angiosperms (Baldwin et al, 1995). Its length was 164 basepairs (bp) in both flowering-time variants as well as in G. odoratissima. The lengths of the ITS 1 and 2 regions were 247 and 240 bp respectively in Gymnadenia spp., as reported for G. conopsea (GenBank Accession no. GCZ94067 and 68). The Gymnadenia sequences formed two distinct major clusters with high support from bootstrap analyses (98 and 99% respectively, based on bootstrap with 1000 replications), with the late-flowering variant and densiflora in one cluster and the early-flowering variant and G. odoratissima with almost identical ITS sequences in another (Figure 3). The differences between the two clusters were due to 12 variable nucleotide sites in ITS 1 and 2: six transitions (three A–G, three C–T) and six transversions (one A–C, one G–C, one G–T, three A–T) (Table 3). Variation within the two clusters was limited.

Consensus of UPGMA trees based on 5.8S RNA, ITS 1 and ITS 2 sequences. Bootstrap percentages are based on 1000 replications, bootstrap support above 50% is given (ie significant support). The tree shows the relationship between early- (E) and late- (L) flowering variants of G. conopsea ssp conopsea, G. conopsea ssp densiflora and G. odoratissima.
Habitat variability
The habitat separation between the early- and late-flowering individuals is illustrated in Figure 4, where the LDA clearly separated the two variants. The early-flowering type also had wider habitat amplitude than the late-flowering type.

Habitat differentiation between late- and early-flowering variants of G. conopsea illustrated by an LDA. The discriminating variables are soil depth, the separate percentage covers of mosses, grasses, Carex and herbs estimated within a circle of 0.10 m radius around each sampled individual and the presence of B. media, E. vaginatum, F. vulgaris, P. lanceolata, P. erecta, S. cearulea, T. montanum and T. repens. The variables that are significantly associated with the flowering-time variants in the GLM analysis are given in the figure at the places along the discriminant axis scale that equal their coefficients of correlation with the axis.
The final GLM had five explanatory variables and an estimate of the random error of 1.18 (dispersion factor). The dispersion is close to the expected unity, and significance testings were made comparing the change in deviance when dropping a term (one at a time) from the final model to a χ2 distribution. The final model uses one degree of freedom each for the five terms, leaving 70 degrees of freedom for the estimation of random error (residual). The change in deviance and corresponding significance level is given in parentheses after each term. The late-flowering type was significantly associated with the presence of E. vaginatum (χ2=6.6, P<0.01) and F. vulgaris (χ2=6.4, P<0.01) and also with higher coverage of grass (χ2=6.7, P<0.01) and herbs (χ2=4.4, P<0.05). The early-flowering variant was associated with the presence of T. montanum (χ2=5.9, P<0.01). The direction of associations was consistent between the LDA and the GLM analyses. However, the LDA was used to illustrate the pattern of differentiation, while conclusions on specific explanatory variables were taken from the GLM. The significance values given in Figure 4 are taken from the GLM analysis.
Discussion
Results from the two types of genetic markers used in this investigation, fast-evolving microsatellite markers and, in comparison, slowly evolving ITS sequences, demonstrated a drastic genetic differentiation and a significant habitat differentiation between early- and late-flowering variants of plants morphologically belonging to G. conopsea ssp conopsea. A few individuals were sequenced for ITS from the close relatives G. conopsea ssp densiflora (late-flowering) and G. odoratissima. There is one closely related group of late-flowering G. conopsea, while the early-flowering G. conopsea are the closest relatives to G. odoratissima.
Habitat differentiation
In populations where the two flowering-time types co-occurred, the early-flowering individuals were significantly associated with the presence of T. montanum, typical of dry grassland, while late-flowering individuals were significantly associated with greater grass and herb cover and with Eriphorum angustifolium, typical for wet habitats. Late-flowering individuals were also significantly associated with the presence of F. vulgaris, which is more difficult to interpret in terms of habitat type. Hence, even within populations, individuals of different flowering types showed different habitat preferences.
Genetic split–historical information
Our study was intended to reveal in detail population genetics and habitat preferences between early- and late-flowering populations that morphologically belong within the ssp conopsea. For comparison, we also included a few samples from morphologically distinct ssp densiflora and from the close relative G. odoratissima. Surprisingly, two flowering-time types were completely separated in the ITS haplotypes by 12 substitutions. Further, the late-flowering type was identical to the late-flowering ssp densiflora, while the early-flowering type was closely related to the morphologically distinct species G. odoratissima (Figure 3). This could indicate that early-flowering populations and the species G. odoratissima have had a historically more recent separation than that of the early- and late-flowering populations of ssp conopsea. Information from more samples and more loci us needed to confirm this finding–gene trees and species trees may not be the same (Nichols, 2001).
Population genetics–recent history
The early-flowering populations were far more genetically diverse and had more alleles per locus than the late-flowering populations (Table 2). The alleles were basically not shared between the flowering-time types, and in three out of four loci alleles diagnostic for the late-flowering could be found. The results can be compared with those of Soliva and Widmer (1999), who discovered strong differentiation between the two subspecies G. conopsea ssp conopsea and ssp densiflora using data from nine allozyme loci. Scacchi and Angelis (1989), 10 years earlier, detected high allozyme divergence among 16 Italian populations of G. conopsea. The populations were divided into two ecotypes, humid and dry. The two ecotypes were differentiated by several fixed alleles, and Scacchi and Angelis (1989) suggested that they might be considered as different species. They claimed that the two ecotypes were morphologically indistinguishable, but they did not study the variation in flowering-time.
We observed some differences in habitat preferences between the flowering types in sympatric populations, perhaps not as pronounced as in Soliva and Widmer (1999) or Scacchi and Angelis (1989), but our results are firm since we measured habitat differentiation between individuals within localities, making the observations independent of large-scale geographic habitat differentiation and giving a good replication of the habitats.
In our study, early-flowering populations were genetically the most variable, showing the highest gene diversity and the largest number of alleles at microsatellite loci, whereas late-flowering populations were comparatively genetically depauperated (Tables 1 and 2). Discriminant analysis (Figure 4) suggested that late-flowering populations have narrower habitat amplitude than early-flowering populations. A possible explanation is that low genetic diversity gives a narrow ecological niche, but this study is not designed to elaborate this issue.
In Soliva and Widmer (1999), ssp conopsea populations were significantly more variable and less differentiated than ssp densiflora. They proposed that the difference could be a consequence of densiflora growing in more moist habitats, habitats that have been strongly reduced over the last century. Consequently, populations of densiflora have been reduced in size and would thereby be more exposed to random genetic drift. This could be a possible explanation also for our late-flowering type; however, if genetic drift in a relatively polymorphic regional population of late-flowering populations alone had created the low genetic diversity, local populations should have a mosaic variation with different alleles fixed (Lönn and Prentice, 1990). This was not the case, since all late-flowering individuals have only a few alleles that were the same in all populations. High levels of selfing might also explain the low local genetic diversity (in this case, in the founding population for Sweden), but inbreeding coefficients are not significantly different between early- and late-flowering populations; so differences in breeding system are less likely to explain the difference in genetic diversity. If the Swedish regional population was founded by a few individuals, founder effect could explain the low diversity in late-flowering populations. Another possible scenario could be that mutation and/or selection resulted in differentiation in flowering-time, creating the late-flowering type. If this event was fairly recent, there would not have been enough evolutionary time for the generation of new genetic variation in microsatellites, hence the observed low genetic variation (see Hultgård, 1987; Brochmann and Elven, 1992). However, such a scenario will not be in accordance with the revealed information from ITS sequences.
Mechanisms of maintenance of the flowering-time variants and future studies
Taken together, information from ITS sequences and microsatellite markers indicated the occurrence of an early historical split between the two phenological conopsea variants, a split that has been maintained until the present time.
There are at least three possible processes or mechanisms that maintain the genetic separation between the early- and late-flowering variants: ecological isolation due to habitat preferences, temporal isolation induced by variation in flowering time and isolation due to different species of pollinators (different sets of Lepidoptera; see Nilsson, 1983). The strong genetic differentiation between the flowering types, occurring in sympatry with very low exchange of genes, suggests a genetic mecha-nism preventing hybrids to cross or back-cross or making hybrid seed less fertile. We also found a few possible hybrids between the flowering-time types, based on diagnostic alleles, whose alleles had not spread within the other flowering-time type populations. The differences in habitat preference and suggested differences in pollinator species (Nilsson, 1983) should then be secondary or have been the factors that once caused the separation of the flowering-time types.
To be able to clarify which genetic processes maintain the separation between the early- and late-flowering types, we need to investigate mixed populations in more detail - screening for hybrids, doing additional cross-pollinations, germinating ‘hybrid’ seed, etc. It is also important from a conservational point of view to examine the actual distribution of mixed and distinct populations and to revise the taxonomic status of the two flowering-time variants. At present, populations of the early-flowering type are endangered due to reduced mowing and cultivation, whereas late-flowering populations and ssp densiflora, which are less dependent on management, slowly increase their acreage. This situation could actually lead to the extinction of the genetically unique early-flowering type, while the species G. conopsea seem to thrive in Sweden. In this case, we would also lose a substantial part of genetic diversification and a large part of the evolutionary potential in the genus Gymnadenia in Sweden.
References
Baldwin BG, Sanderson MJ, Porter MJ, Wojciechowski MF, Campbell CS, Donoghue MJ (1995). The ITS region of nuclear ribosomal DNA: a valuable source of evidence on angiosperm phylogeny. Ann Miss Bot Gard 82: 247–277.
Brochmann C, Elven R (1992). Ecological and genetic consequences of polyploidy in arctic Draba (Brassicaceae). Evol Trends Plants 6: 111–124.
Crawley MJ (1993). GLIM for Ecologists. Blackwell; London.
Gustafsson S, Thorén P (2000). Characterization of microsatellite loci in the fragrant orchid, Gymnadenia conopsea. Mol Ecol Notes 1: A7.
Hauser TP, Weidema IR (2000). Extreme variation in flowering time between populations of Silene nutans. Hereditas 132: 95–101
Heusser C . (1938). Chromosomenverhältnisse bei schweizerischen basitonen Orchideen. Ber Schweiz Bot Ges 48: 562–605.
Hultga˚rd U-M (1987). Parnassia palustris L. in Scandinavia. Acta Univ Ups Symb Bot Ups 18: 1–128
Karlsson T (1984). Early-flowering taxa of Euphrasia (Schrophulariaceae) on Gotland, Sweden. Nord J Bot 4: 303–326.
Krok Th, Almquist S (1994). Svensk Flora, 27th edn. Almqvist & Wiksell Tryckeri: Uppsala.
Kumar S, Tamura K, Jakobsen IB, Nei M (2001). MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17: 1244–1245.
Lennartsson T (1997). Seasonal differentiation–a conservative reproductive barrier in two grassland Gentianella (Gentianaceae) species. Plant Syst Evol 208: 45–69.
Lenormand T (2002). Gene flow and the limits to natural selection. Trends Ecol Evol 17: 183–189.
Leskinen E, Pamilo P (1997). Evolution of the ITS sequences of ribosomal DNA in Enteromorpha (Chlorophyceae). Hereditas 126: 17–23.
Levin DA (2001). 50 years of plant speciation. Taxon 50: 69–91.
Li CC (1976). First Course in Population Genetics. The Boxwood Press; California.
Lönn M, Prentice HC (1990). Mosaic variation in Swedish Petrorhagia prolifera (Caryophyllaceae): the partitioning of morphometric and electrophoretic diversity. Biol J Linn Soc 41: 353–373.
MathSoft (1999). S-PLUS 2000 User's Guide. Seattle, WA.
Mayr E (1970). Population, Species and Evolution. Harward University Press: Cambridge, MA.
Mossberg B, Stenbeck L (1992). Den nordiska floran, 3rd edn. Wahlström & Widstrand: Turnhout.
Neuffer B, Hurka H (1999). Colonization history and introduction dynamics of Capsella bursa-pastoris (Brassicaceae) in North America: isozymes and quantitative traits. Mol Ecol 8: 1667–1681.
Nichols R (2001). Gene trees and species trees are not the same. Trends Ecol Evol 16: 358–364.
Nilsson LA (1983). I orkidéernas förtrollade värld. In: Gillsäter S, Malmberg S (eds) Öland–nattviol och näktergal, Trevi: Stockholm. pp 121–138.
Prentice HC, Cramer W (1990). The plant community as a niche bioassay: environmental correlates of local variation in Gypsophila fastigiata. J Ecol 78: 313–325.
Proctor M, Yeo P, Lack A (1996). The Natural History of Pollination. Harper Collins Publishers: London.
Raymond M, Rousset F (1995). Genepop (Version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Rundle HD, Whitlock MC (2001). A genetic interpretation of ecologically dependent isolation. Evolution 55: 198–201.
Scacchi R, Angelis GD (1989). Isoenzyme polymorphism in Gymnadenia conopsea and its inferences for systematics within this species. Biochem Syst Ecol 17: 25–33.
Slatkin M (1987). Gene flow and geographic structure of natural populations. Science 236: 787–792.
Soliva M, Widmer A (1999). Genetic and floral divergence among sympatric populations of Gymnadenia conopsea SL (Orchidaceae) with different flowering phenology. Int J Plant Sci 160: 897–905.
Turelli M, Barton NH, Coyne JA (2001). Theory and speciation. Trends Ecol Evol 16: 330–343.
Van Dijk H, Boudry P, McCombie H, Vernet P (1997). Flowering time in wild beet (Beta vulgaris ssp maritima) along a latitudinal cline. Acta Oecol 18: 47–60.
Weir B, Cockerham C (1984). Estimating F-statistics for the analyses of population structure. Evolution 38: 1358–1370.
Venables WN, Ripley BD (1999). Modern Applied Statistics with S-Plus, 3rd edn. Springer: New York.
Acknowledgements
This study was funded by The Oscar and Lili Lamm foundation, Nilsson-Ehle foundation (to SG) and by Helge Ax:son Johnsons stiftelse (to ML). We are very grateful to Martin Lascoux, Anders L Nilsson, Pekka Pamilo, Per Sjögren-Gulve and Peter Thorén for their helpful comments on the earlier versions of the manuscript, and to Måns Stefansson for help in field work. We also thank many Botanical Societies, Bo Göran Johansson, Karin Bengtsson, Barbro Risberg, Mikael Hedrén and Anna-Lena Fritz for providing very important information about the localities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gustafsson, S., Lönn, M. Genetic differentiation and habitat preference of flowering-time variants within Gymnadenia conopsea. Heredity 91, 284–292 (2003). https://doi.org/10.1038/sj.hdy.6800334
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800334
Keywords
This article is cited by
-
Molecular and morphological phylogenetics of the digitate-tubered clade within subtribe Orchidinae s.s. (Orchidaceae: Orchideae)
Kew Bulletin (2018)
-
Genetic variability and population structure in a collection of inbred lines derived from a core germplasm of castor
Journal of Plant Biochemistry and Biotechnology (2017)
-
Effects of population structure on pollen flow, clonality rates and reproductive success in fragmented Serapias lingua populations
BMC Plant Biology (2015)
-
Exploring the impact of neutral evolution on intrapopulation genetic differentiation in functional traits in a long-lived plant
Tree Genetics & Genomes (2014)
-
Strong genetic differentiation between Gymnadenia conopsea and G. densiflora despite morphological similarity
Plant Systematics and Evolution (2011)
