
Abstract
Background
Leber's congenital amaurosis (LCA) is an inherited retinal dystrophy, which causes severe visual impairment in early childhood. Recent molecular genetic studies have linked 11 loci (AIPL1, CRB1, CRX, GUCY2D, RPE65, RDH12, RPGRIP1, TULP1, LCA3, LCA5, and LCA9) to LCA. LCA5 is a new locus, which maps to the 6q11–q16 chromosomal region and was found to be associated with macular coloboma-type LCA in a Pakistani family. Herein, we describe the molecular genetic features of a consanguineous Turkish family in which four children have macular coloboma-type LCA.
Methods
Haplotype analysis was performed on the DNA of the family members using microsatellite markers against GUCY2D, RPE65, and LCA5. Genomic DNA was screened for mutations by means of single-strand conformational polymorphism (SSCP) analysis in exons of the RPE65 and CRX genes.
Results
In haplotype analysis, no linkage to LCA5 or GUCY2D loci was detected. None of the tested markers showed homozygosity or segregation between affected siblings. PCR-SSCP mutation analysis revealed no mutations in the screened RPE65 and CRX genes.
Conclusion
We excluded LCA5 as the genetic cause of macular coloboma-type LCA in this Turkish family. Macular coloboma-type LCA shows genetic heterogeneity and it is not possible to establish a phenotype–genotype correlation with LCA5 and macular coloboma.
Similar content being viewed by others

Introduction
Leber's congenital amaurosis (LCA, MIM 204000) is an inherited autosomal recessive retinal dystrophy that leads to severe visual impairment in early childhood. Clinical features of LCA are low vision or blindness, nystagmus, eye rubbing, photophobia, and the absence of, or severely reduced, electroretinogram (ERG). The retinal appearance may be normal early in the course of the disease, but eventually shows changes similar to retinitis pigmentosa, such as bone spicule pigmentation and optic nerve pallor. Yellow flecks can be seen in the equatorial fundus. It can also present with bull's eye-type macular changes and macular coloboma-like lesions.1
Genetic defects in GUCY2D (retinal guanylate cyclase, 17p13.1),2 AIPL1 (aryl hydrocarbon receptor-interacting protein-like 1, 17p13.2),3 CRB1 (Crumbs homolog 1, 1q31.3),4 CRX (photoreceptor specific cone–rod homeobox transcription factor, 19q13.32),5 RPE65 (retinal pigment epithelium specific 65 kDa protein, 1p31.2),6 RPGRIP1 (retinitis pigmentosa GTPase regulator-interacting protein 1, 14q11.2),7 RDH12 (retinal dehydrogenase 12, 14q24.1)8 and TULP1 (Tubby-like protein 1, 6p21.31)9 have been associated with LCA. Additionally, three independent loci with unidentified genes, LCA3 (14q24),10 LCA5 (6q11–q16),11 and LCA9 (1p36)12 have been found to show linkage to LCA. LCA5, which maps to the 6q11–q16 chromosomal region, was found to be associated with macular coloboma-type LCA in a consanguineous Pakistani family.13 Herein, we describe the molecular genetic features of a consanguineous Turkish family in which four children have macular coloboma-type LCA.
Patients and methods
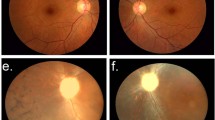
The clinical features of the three brothers in this family were described in our previous report.14 In brief, these children aged 13, 11, and 9 years, respectively, complaining of visual difficulties in the dark, had macular coloboma-type LCA. Their best-corrected visual acuities ranged between 20/100 and 20/25. All had nonrecordable ERGs. During 5 years of follow-up, the third child had deterioration of his vision and slight enlargement of the macular lesions. The other two brothers remained stable. Their sister was first examined in 2003 when she was 6 months old. The fundus examination was normal. After 1 year, her parents reported that she had trouble seeing and she had roving eye movements. In ophthalmic examination, a relatively well-circumscribed bilateral macular atrophy, yellow flecks, and pigmentation in the fundus, very similar to her brothers, were seen (Figure 1a and b). All four siblings' ERGs were nonrecordable. The children had no associated systemic disorders. The parents were second cousins and had no family history of severe visual impairment or blindness. Their ophthalmic findings were normal.

Fundus photographs of the right eye (a) and left eye (b) of the sister, at 2 years of age, showing bilateral macular coloboma.
Venous blood samples were obtained from all the participants for DNA extraction and genomic DNA was isolated according to the standard methods. Informed consents were obtained from the parents and the study received approval from the Ethical Review Board of Hacettepe University.
Haplotype analysis and PCR-SSCP
For haplotype analysis, microsatellite markers were chosen from the chromosomal regions that contain the candidate loci, GUCY2D, RPE65, and LCA5. Tightly linked or closely located markers were amplified using family members' DNA. All exonic sequences and exon–intron boundaries of CRX and RPE65 genes were screened by PCR-SSCP. Primer and marker sequences for the PCR were selected from the Ensembl Genome Database (http://www.ensembl.org). The PCR product was routinely run in a 2% agarose gel to check the DNA purity. After amplification, PCR products were mixed with formamide dye and denatured for 3 min at 95°C. Denatured samples were loaded on 7% denaturing polyacrylamide gels for genotyping. For SSCP analysis, samples were run in nondenaturing polyacrylamide gels. After 4–5 h at 1600 V electrophoresis, the gels were stained with silver nitrate and photographed for genotyping.
Results
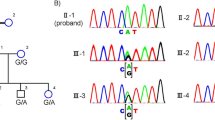
None of the tested markers showed homozygosity or segregation between affected siblings for LCA5 (Figure 2), GUCY2D, and RPE65 loci. PCR-SSCP mutation analysis revealed no mutations in the screened RPE65 and CRX genes.

Pedigree of our consanguineous Turkish family with macular coloboma-type LCA and haplotype of markers in the LCA5 chromosomal region.
Discussion
LCA constitutes 5% of all hereditary retinal dystrophies and has a higher incidence where consanguineous marriages are common.15 Fundus findings are extremely variable ranging from normal to widespread peripheral or macular retinal degenerations.1, 15 The genetic heterogeneity of LCA might affect the disease phenotype and lead to different clinical presentations. In the series of Perrault et al,16 GUCY2D, RPE65, and CRX genes accounted for 27% of Leber cases. Hanein et al,9 screened seven LCA causative genes in 179 unrelated LCA cases and found mutations in 47.5% of the patients (GUCY2D (21.2%), CRB1 (10%), RPE65 (6.1%), RPGRIP1 (4.5%), AIPL1 (3.4%), TULP1 (1.7%), and CRX (0.6%)). It is obvious that there are still unidentified mutations in LCA and further genetic mapping and cloning of new candidate genes are required.9 In another genetic study,17 a homozygous nonsense mutation, Trp278X, in the photoreceptor-pineal gene AIPL1 was found in four Pakistani families with LCA. These patients presented with a severe phenotype of LCA with hand motion to no light perception vision and fundus findings ranging from maculopathy to diffuse pigmentary retinopathy. LCA with CRB1 mutations can present with white spots and pigment clumps with moderate to high hyperopia.4 Mutations in GUCY2D and RPE65 genes are associated with normal fundus findings at birth, followed by salt and pepper appearance with varying functional outcomes.16
In this report, we described the clinical features of a Turkish family with macular coloboma-type LCA. The genetic analysis showed a lack of linkage to LCA5 locus in contrast to a report by Mohamed et al.13 The affected individuals from both families presented with bilateral macular atrophy and perifoveal pigmentary changes. They all had normal neurological examinations and intelligence. However, macular changes seemed to begin at an earlier age in the members of our Turkish family and their visual acuities were better. In our family, we have also excluded mutations in the known LCA genes, namely GUCY2D, RPE65, and CRX. Macular coloboma-type of LCA shows genetic heterogeneity and it is not possible to establish a phenotype–genotype correlation with LCA5 and macular coloboma.
References
Smith D, Oestreicher J, Musarella MA . Clinical spectrum of leber's congenital amaurosis in the second to fourth decades of life. Ophthalmology 1990; 97: 1156–1161.
Perrault I, Rozet JM, Calvas P, Gerber S, Camuzat A, Dollfus H et al. Retinal specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat Genet 1996; 14: 461–464.
Sohocki MM, Bowne SJ, Sullivan LS, Blackshaw S, Cepko CL, Payne AM et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet 2000; 24: 79–83.
Lotery AJ, Jacobson SG, Fishman GA, Weleber RG, Fulton AB, Namperumalsamy P et al. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol 2001; 119: 415–420.
Freund CL, Wang QL, Chen S, Muskat BL, Wiles CD, Sheffield VC et al. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet 1998; 18: 311–312.
Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C et al. Mutations in RPE65 cause Leber's congenital amaurosis. Nat Genet 1997; 17: 139–141.
Dryja TP, Adams SM, Grimsby JL, McGee TL, Hong DH, Li T et al. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet 2001; 68: 1295–1298.
Perrault I, Hanein S, Gerber S, Barbet F, Ducroq D, Dollfus H et al. Retinal dehydrogenase 12 (RDH12) mutations in Leber congenital amaurosis. Am J Hum Genet 2004; 75: 639–646.
Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype–phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 2004; 23: 306–317.
Stockton DW, Lewis RA, Abboud EB, Al-Rajhi A, Jabak M, Anderson KL et al. A novel locus for Leber congenital amaurosis on chromosome 14q24. Hum Genet 1998; 103: 328–333.
Dharmaraj S, Li Y, Robitaille JM, Silva E, Zhu D, Mitchell TN et al. A novel locus for Leber congenital amaurosis on chromosome 6q. Am J Hum Genet 2000; 66: 319–326.
Keen TJ, Mohamed MD, McKibbin M, Rashid Y, Jafri H, Maumenee IH et al. Identification of a locus (LCA9) for Leber's congenital amaurosis on chromosome 1p36. Eur J Hum Genet 2003; 11 (5): 420–423.
Mohamed MD, Topping NC, Jafri H, Raashed Y, McKibbin MA, Inglehearn CF . Progression of phenotype in Leber's congenital amaurosis with a mutation at the LCA5 locus. Br J Ophthalmol 2003; 87: 473–475.
Kıratlı H, Bozkurt B . Macular Coloboma Type Leber's Congenital Amaurosis [Artıcle In French]. J Fr Ophtalmol 2002; 25: 67–70.
Hanein S, Perrault I, Gerber S, Tanguy G, Hamel C, Dufier JL et al. Amaurose congénitale de Leber: le point sur l'hétérogénéité génétique, actualisation de la définition clinique. J Fr Ophtalmol 2005; 28: 98–105.
Perrault I, Rozet JM, Gerber S, Ghazi I, Leowski C, Ducroq D et al. Leber congenital amaurosis. Mol Genet Metab 1999; 68: 200–208.
Damji KF, Sohocki MM, Khan R, Gupta SK, Rahim M, Loyer M et al. Leber's congenital amaurosis with anterior keratoconus in Pakistani families is caused by the Trp278X mutation in the AIPL1 gene on 17p. Can J Ophthalmol 2001; 36: 252–259.
Acknowledgements
We thank the members of the family for their participation in this study. This study was supported by the TÜBİTAK BAYG-BDP and DPT 01K120660.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Özgül, R., Bozkurt, B., Kıratlı, H. et al. Exclusion of LCA5 locus in a consanguineous Turkish family with macular coloboma-type LCA. Eye 20, 817–819 (2006). https://doi.org/10.1038/sj.eye.6702024
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6702024
