
Abstract
Background:
We have recently reported an inverse relationship between colon cancer progression and tumour proliferative activity. Here, we extend our findings by evaluating the proliferative activity of liver metastatic lesions and primary colorectal cancers (CRC) that differ in their metastatic potential.
Methods:
Using an earlier established multi-gene proliferation signature (GPS), proliferative levels were analysed in 73 primary CRCs and 27 liver metastases.
Results:
Compared with primary CRCs, we observed a significantly lower expression of the GPS in liver metastases and confirmed their lower proliferative levels by quantitative RT–PCR and Ki-67 immunostaining. No difference could be detected in apoptotic indices as assessed by M30 immunostaining, indicating that the net growth rate is lower in metastases relative to primary tumours. Notably, relapsed primaries or those with established metastases had significantly lower proliferative activity than CRCs that were non-metastatic and did not relapse.
Conclusion:
Our results suggest that slow proliferation is a biological characteristic of both liver metastases and those primary tumours with the ability to metastasise. The delineation of the mechanisms underlying the inverse association between proliferation and CRC aggressiveness may be important for the development of new therapeutic strategies.
Similar content being viewed by others


Main
Metastasis is the major cause of cancer death. The underlying process consists of a series of interrelated biological steps that eventually lead to the formation of metastatic lesions. This highly selective process favours the survival and proliferation of a sub-population of cancer cells with the ability to complete all steps. In addition to these intrinsic features of metastatic cells, their interaction with the host organ is another potential determinant of successful metastasis formation (Steeg, 2006). However, our knowledge of the mechanisms driving the spread of primary tumours is limited.
Colorectal cancer (CRC) is one of the most common causes of cancer-related mortality in the Western world and metastasis to the liver is responsible for a large proportion of the deaths from CRC. Approximately 50% of CRC patients develop liver metastases during the course of their disease (Bird et al, 2006). Although surgery and adjuvant therapy can effectively control CRCs at the primary site, 80–90% of metastatic lesions are not resectable (Adam and Vinet, 2004). Even with access to modern chemotherapy drugs, non-operable patients have a median survival rate of less than 2 years (Wolpin and Mayer, 2008). A deeper understanding of the biology of metastasis may improve the ability to identify patients at high risk of developing metastases and lead to the discovery of better treatments for established metastatic disease.
A fundamental feature of cancer cells is their altered proliferative rate, which is often used as a basis for current chemotherapies. We, however, have recently reported an inverse relationship between the proliferative activity of primary colon cancers and their malignant potential by utilising a multi-gene proliferation signature (GPS) (Anjomshoaa et al, 2008). The GPS provides an objective and robust assay for the assessment of proliferation, as its expression is increased relative to quiescent cells in both (i) proliferating CRC cells in vitro and (ii) the proliferative region of colonic crypts in vivo. The GPS was tested on breast cancer, where it correctly identified the well-established association between increased proliferation and worse outcome. However, when the GPS was used to analyse biopsy material from two independent cohorts of colon cancer patients, lower cellular proliferation was found to be associated with advanced disease stages and a shorter disease-free survival. Of note, these associations were independent of adjuvant chemotherapy received by some patients.
Here, we present evidence corroborating the association between more aggressive biological behaviour and low proliferation in CRC by analysing CRC liver metastases and a series of primary tumours differing in metastatic potential. We found that both metastasising primaries and liver metastases are characterised by a reduced proliferative activity relative to non-metastatic CRCs.
Materials and methods
Patients and tumour samples
Tumour specimens were obtained from 73 patients undergoing surgery for primary CRC at Dunedin Public Hospital and 27 patients undergoing resection of CRC liver metastases at Auckland City Hospital, New Zealand. Fresh frozen specimens were taken from each tumour and stored at −80°C. Formalin-fixed paraffin-embedded tissue blocks were also available. Ethical Committee approval was obtained and all participants gave signed informed consent.
The primary tumours and liver metastases were not paired. Of the 27 patients operated on for liver metastases, 10 had synchronous and 17 had metachronous disease. Of the 73 primary tumours, 40 were situated in the colon and 33 were rectosigmoid or rectal. Ten had stage I, 27 stage II, 28 stage III and 8 stage IV disease. Patients were followed up for a minimum of 5 years. Of the 65 patients with primary CRC who underwent potentially curative resection (stage I–III patients), 24 developed disease relapse, 21 of which included systemic spread. Three isolated local relapses were not included in the analyses.
Array preparation and gene expression analysis
Gene expression profiling of all tumours was performed using arrays spotted with an MWG 30K Oligo Set (MWG Biotech, Winston-Salem, NC, USA) as described previously (Anjomshoaa et al, 2008). Briefly, Cy dyes were incorporated into cDNAs synthesised from 10 μg tumour sample or reference RNA using the indirect amino-allyl cDNA labelling method. A pooled RNA sample derived from a mixture of 12 cell lines was used as a reference for all hybridisations. The mixture of dye-labelled cDNAs was then purified and co-hybridised to a microarray. After scanning with a GenePix 4000B microarray scanner (Molecular devices, Sunnyvale, CA, USA), the foreground intensities from each channel were log2-transformed and normalised using the SNOMAD software (Colantuoni et al, 2002). Normalised values were collated and filtered using BRB-Array Tools version 3.6.0-β_3 (http://linus.nci.nih.gov/BRB-ArrayTools.html). Genes of low signal intensity, or for which more than 20% of measurements across tumour samples were missing, were excluded from further analysis.
Quantitative real-time–PCR
The expression of seven randomly selected genes from the GPS (MAD2L1, POLE2, CDC2, MCM6, MCM3, PBK and GMNN) and two well-known proliferative genes (PCNA and Ki-67) was validated by quantitative real-time–PCR (qRT–PCR) on an ABI Prism 7900HT system, Applied Biosystems, Foster City, NC, USA. Relative fold changes were calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001). Topoisomerase 3A (TOP3A) was used as the internal control because it had the least expression variation across samples compared with other potential housekeeping genes (i.e. ACTB, DSP, BAZ2A). Reference RNA (the same reference as for microarrays) was used as a calibrator to enable comparison between different experiments.
Immunohistochemistry
Immunohistochemical assessment of proliferation and apoptosis was performed in all tumours. Antigens were retrieved on 4 μm sections in boiling citrate buffer (pH 6). Primary antibodies (MIB-1/Ki-67 and M30 Cytodeath; dilution 1 : 50; Dako Glostrup, Denmark) were detected using the EnVision system (Dako) and the DAB substrate kit (Vector Laboratories, Burlingame, CA, USA). For each tumour sample, cells within five high-power fields were counted by two observers in a blinded manner. The proliferative and apoptotic indices were presented as percentage of positively stained cells.
Statistical analysis
All microarray analyses were performed using BRB-Array Tools software. Testing for significant differences in the GPS expression or immunohistochemical indices between groups of tumours was carried out with SPSS 15.0.0 (SPSS Inc., Chicago, IL, USA). A multiple comparisons correction (Bonferroni) was applied to the P-values derived from testing for differences in microarray-based gene expression. Additional details relating to the statistical methods and their specified parameters are described in the corresponding results sections.
Results
Lower GPS expression in liver metastases compared with primary CRCs
To investigate whether liver metastases and primary CRCs differ in their proliferative activity, the expression of genes constituting our GPS was compared between the two tumour groups. Expression values for the GPS genes were obtained from the array-generated expression profiles. Expression data were available for 33 out of the 36 GPS genes. CDCA5, KIF4A and GINS2 were filtered out due to the poor quality of spots. We first examined whether GPS expression differs between tumours of rectal and colonic origin. Because the GPS levels were similar in the rectal and colonic groups (adjusted P>0.4, two-tailed t-test based on 1000 permutations), rectal and colon tumours were considered as one group in all subsequent analyses. Intriguingly, 30 out of the 33 GPS genes had significantly lower expression in liver metastases compared with primary CRCs (adjusted P<0.01; two-tailed t-test) (Supplementary Table 1). Only three genes (CDC20, NOLA2 and SSX2IP) had similar expression levels. These results suggest that liver metastases are less proliferative than primary CRCs.
Lower expression by qRT–PCR of GPS genes in liver metastases compared with primary CRCs
To validate the microarray data, seven genes from the GPS (MAD2L1, POLE2, CDC2, MCM6, MCM3, PBK and GMNN) and two established proliferation-related genes (PCNA and Ki-67) were assessed by qRT–PCR. As shown in Figure 1, all examined genes displayed significantly lower expression in liver metastases relative to primary CRCs (P<0.005; Mann–Whitney U-test). Strong (correlation coefficients 0.74–0.86) and highly significant (P<0.001; Spearman's correlation test) correlation was observed between the microarray- and qRT–PCR-generated expression data.

Quantitative RT–PCR confirmed the lower expression of nine cell-cycle-related genes in liver metastases compared with primary CRCs. The Y axis represents the fold changes calculated as the ratio of tumour to reference gene expression, normalised to a control gene. The box range (interquarter range) contains the middle 50% of the data. The median value is shown as a horizontal line across the box. The extreme values (within 1.5 times the interquartile range from the upper or lower hinges) are the ends of the whiskers. Points at a greater distance than 1.5 and 3 times of the interquartile range are outliers and presented as circles and asterisks, respectively. All differences were significant at the α-level of 0.005 using the Mann–Whitney U-test. Analysis was performed using SPSS software.
Lower Ki-67 labelling index in liver metastases compared with primary CRCs
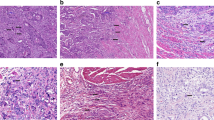
To determine whether liver metastases and primary CRCs differ in Ki-67 nuclear expression, the Ki-67 labelling index (LI) was compared between the two groups (Figure 2A and B). Median (range) Ki-67 LI was 81.8% (25.1–96.4%) in primary CRCs, and 36.2% (12.1–65.5%) in liver metastases (P<0.001; Mann–Whitney U-test; Figure 2C).

Nuclear staining of Ki-67 protein in a primary CRC (A) and a liver metastasis (B). Cytoplasmic staining of M30 apoptotic protein in a primary CRC (D) and a liver metastasis (E). The Ki-67 LI was significantly lower in liver metastases compared with primary CRCs (C). No significant difference was observed in M30 AI between liver metastases and primary CRCs (F). P-values were calculated using the Mann–Whitney U-test.
Similar M30 apoptotic index in liver metastases and primary CRCs
To determine whether liver metastases and primary CRCs differ in apoptotic rates, the M30 apoptotic index (AI) was assessed in the two groups (Figure 2D and E). Median (range) M30 AI was 4.28% (0.83–17.95%) in primary CRCs and 3.91% (1.45–11.2%) in liver metastases. No significant difference in the M30 AI was detected between primary CRCs and liver metastases (P=0.68; Mann–Whitney U-test; Figure 2F). The observation of lower proliferative index of liver metastases relative to primaries, together with a similar AI, indicates that the net growth rate is lower in liver metastases than in primary CRCs.
Slow proliferation is a biological feature of CRC metastasis
Because metastatic lesions are the result of a process that is initiated at the primary tumour site, they should share biological features with primary tumours that have a high metastatic potential. We therefore hypothesised that primary CRCs with a high metastatic potential have slower proliferative rates than CRCs with a low metastatic potential. To test this hypothesis, GPS genes were first averaged to assign each tumour a mean GPS score. GPS scores were then compared between groups of tumours classified according to the tumour stage or relapse status. P-values were calculated using the Exact test option in SPSS, which provides more accurate estimates of significance for non-parametric tests when dealing with small sample sizes. The Mann–Whitney U-test was used for comparison of two groups and Kruskal–Wallis test for comparison of more than two groups. The results are summarised below:
-
a)
To determine whether tumour proliferation is related to the presence of metastasis at the time of surgery, GPS scores were compared between stage I–II and stage III–IV primary tumours. Stage I and II CRCs were grouped as they were confined to the intestinal wall and no difference in proliferation was detected between non-relapsed stage II and I tumours (P=0.77). Stage III and IV CRCs were grouped because these two stages of disease did not differ in proliferation (P=0.92). Stage III–IV tumours had significantly lower GPS scores than stage I–II tumours (P=0.022; Figure 3A). This analysis suggests that CRCs with established metastasis to the lymph nodes or distant organs have lower proliferative activity than CRCs that were non-metastatic at the time of surgery.
Figure 3 
Box plots summarising the distribution and variability of GPS scores in defined classes of CRCs. GPS expressions for 33 genes (log2 target/reference) were first averaged for each sample, and then transformed into fold changes as GPS scores. The exact levels of significance were calculated using the Exact method implemented in Mann–Whitney U-test (for comparison of two groups) and Kruskal–Wallis test (for comparison of more than two groups). Resulting P-values are indicated in the right upper part of each panel. Numbers in the upper left of plots refer to the corresponding paragraphs in the manuscript. The interpretation of box plots has been described in the legend of Figure 1.
-
b)
To determine whether slow proliferation in stage I–II tumours was associated with disease relapse, GPS scores were compared between the tumours of patients who remained relapse-free and those who relapsed. All stage I tumours remained relapse-free, while 7 out of 27 stage II tumours experienced relapse during a 5-year follow-up. The GPS scores were found to be significantly lower in the relapsed compared with the non-relapsed group (P=0.014; Figure 3B). When analysis was limited to stage II tumours, the association between tumour proliferation and relapse remained significant (P=0.035).
-
c)
To determine whether proliferation is associated with the likelihood of relapse in tumours with lymph node metastasis at the time of surgery, only stage III CRCs were analysed. No difference in the GPS scores was detected between 11 relapse-free tumours and 14 tumours that relapsed after surgery (P=0.44; Figure 3C). Three tumours had local relapse and were not included in the analysis. Stage IV tumours were not analysed as all had residual disease from the time of surgery. These results suggest that low proliferation is not associated with the likelihood of relapse but rather with the ability to disseminate beyond the primary site.
-
d)
To determine whether slow proliferation is a common feature of primary tumours with metastasis at the time of surgery and metastasis during follow-up, the GPS scores of stage III–IV and stage II CRCs that relapsed during follow-up was compared. Interestingly, no difference in the GPS scores was detected (P=0.78; Figure 3D), indicating that slow proliferation is part of a metastatic signature common to both. These results confirm that slow proliferation is associated with the ability to metastasise as assessed under (C).
-
e)
To further confirm the association between slow proliferation and metastatic potential in primary tumours, relapse-free stage I–II tumours and stage III–IV tumours were analysed. As expected, GPS scores were significantly lower in stage III–IV tumours compared with the other group (P=0.013; Figure 3E).
-
f)
To determine whether CRC proliferative activity inversely correlates with increasing metastatic potential, we compared GPS scores between three groups of tumours. Group 1 (non-metastatic CRCs) included stage I–II tumours from patients who remained relapse-free. Group 2 (metastatic CRCs) included stage III–IV and stage II tumours from patients who developed relapse. Group 3 included the liver metastases. GPS scores were significantly different between the three groups (P=0.001; Figure 3F). Proliferative activity increased from liver metastases to metastatic primaries and was highest in non-metastatic primaries, confirming that slow proliferation is related to the ability of CRCs to metastasise.

Gene set comparison analysis confirms the significant differences in the GPS between tumour groups differing in their metastatic potential
To confirm the significant differences in proliferative activity of the tumour groups defined above (A–F), we used gene set comparison analysis as an alternative approach. When comparing different tumour groups, the GPS was considered to have a higher than expected number of differentially expressed genes if the Kolmogorov–Smirnov (KS) re-sampling P-value was less than 0.005 (default value). The distribution of KS statistics was obtained following 100 000 random re-sampling events. Gene set comparison analysis confirmed that the GPS contained a higher than random proportion of DE genes among groups as was observed with the non-parametric tests (data not shown).
Altogether, the comparison of GPS levels in the various CRC groups strongly suggests that slow proliferation is a biological feature of CRCs that have the potential to metastasise.
Discussion
Dysregulated cellular proliferation is a defining characteristic of cancer. It is therefore not surprising that cell proliferation has received considerable attention in the field of cancer biology and has been extensively studied as a means to predict the behaviour of tumours. The general perception says that a selective growth advantage is fundamental to the development and progression of cancers. Many studies support this concept by demonstrating that more aggressive tumours have a higher proliferative capacity (Rosenwald et al, 2003; Krasnoselsky et al, 2004; Dai et al, 2005). However, we have recently reported that the opposite is true in colon cancer (Anjomshoaa et al, 2008).
In the present study, the significantly lower proliferative activity of liver metastases relative to primary CRCs provides additional evidence for a correlation between slow proliferation and high aggressiveness of CRC. Our conclusion is based on the differential expression of multiple proliferation-associated genes (the GPS) between liver metastases and primary CRCs, which was confirmed by single-gene quantitative RT–PCR assays. Furthermore, Ki-67 immunostaining confirmed the low levels of proliferation in CRC metastases and demonstrated that the difference in the proliferative activity between liver metastases and primary CRCs was large enough to be detected by a subjective single-protein assay. Finally, the combination of a lower proliferative activity with a similar AI in metastases relative to primaries is consistent with the clinical observation that liver metastases are sometimes slow growing. Other groups using immunohistochemical methods have come to similar conclusions (Thomas et al, 1998; Agui et al, 2002; Kitabatake et al, 2002; Seong et al, 2004).
An important observation in our study is that low relative proliferation appears to be a biological feature of CRCs with a high metastatic potential. This viewpoint is strongly supported by the GPS-based analysis of primary CRCs, where groups of highly malignant tumours (high stage and relapse) displayed low proliferative activity relative to less malignant tumours (low stage and low risk of relapse). All observed associations between metastatic capacity of primary CRCs and their proliferative activity also hold up in the cohort of 108 tumours analysed in our earlier study (Anjomshoaa et al, 2008). The low proliferation of metastatic CRCs was intermediate between that of non-metastatic CRCs and liver metastases, further strengthening this view. This is consistent with the expression profiling studies that have demonstrated a substantial signature overlap between metastases and their original primaries, but a lack thereof between metastatic and non-metastatic primaries (Koehler et al, 2004; D’Arrigo et al, 2005). We therefore propose that reduced proliferative activity is part of a biological signature that renders CRCs aggressive.
There are some strengths and weaknesses in this study. Strengths include the multi-gene method of assessing proliferation, which has been validated earlier (Anjomshoaa et al, 2008) and was further validated in this study by RT–PCR and an established immunohistochemical method for the measurement of proliferation. Another strength is the long and complete follow-up in our cohort of patients with primary CRC, which minimises the risk of mis-classification when separating the groups according to relapse. A weakness of the study is that the 27 patients with liver metastases all underwent liver resection, yet the majority of patients with liver metastases from CRC are not resectable. This cohort may thus not be representative of the broader group with liver metastases. This limitation was imposed because it is difficult to obtain fresh tumour samples from patients with unresectable liver metastases. Nonetheless, it seems unlikely that the biological behaviour of resectable liver metastases will significantly differ from that of non-resectable ones with respect to the relative proliferation levels. Furthermore, the fact that the primaries and metastatic lesions were not matched limits the assessment of the relative proliferation differences. In addition, although we could not detect any difference in proliferation between tumours with metastasis to the lymph nodes and those with distant organ metastasis, this result remains speculative because of the small numbers of stage IV tumours.
Given that metastasis is a highly selective process and distant metastases arise from a small sub-population of aggressive cells within the heterogeneous primary lesion, it is conceivable that the biological features of the original cells will manifest themselves in the metastatic lesion. If the cells of origin are slow proliferating and resistant to apoptosis, the cells at the metastatic sites are likely to be as well. Consistent with this scenario are the intermediate proliferative levels of metastatic primaries (a sub-population of slow-growing cells within a heterogeneous tumour) and the low levels of variation among the GPS genes in the metastases (where a single invading cell has produced a clone at a distant site; see also Figure 1). In this context, the ability to survive in the secondary organ may dominate the need to proliferate fast. However, inhibitory growth signals and an inappropriate (non-physiological) environment at the host organ site may also contribute to the slow proliferation of liver metastases (Hara et al, 2000).
There are a number of plausible mechanisms that could underlie the association between low cellular proliferation and disease progression in CRC. Although it is intuitively logical that accelerated cell division allows for more genetic errors to accumulate and to promote tumour progression, slow proliferation can be a feature of cancer cells with the ability to spread. For example, the presence of dissociated, dedifferentiated tumour cells (budding cells) at the tumour–host interface correlates with an enhanced malignant potential in CRC (Prall, 2007). It is noteworthy that budding cells have undergone an epithelial–mesenchymal transition (EMT) and are growth arrested due to increased expression of the cell-cycle-dependent kinase inhibitor, P16 and other unknown mechanisms (Jung et al, 2001). Furthermore, low cellular proliferation at the invasive margin has been associated with poor prognosis in CRC (Palmqvist et al, 1999). Therefore, CRCs with a high proportion of dedifferentiated cells or with an extensive tumour–host interface may be more slowly proliferating but aggressive.
Tumour cells at the tumour–host interface not only express EMT-associated genes, but also increase the expression of stemness-associated genes and therefore are considered to be migratory cancer stem cells (Brabletz et al, 2005). Cancer stem cells are relatively quiescent (Boman et al, 2008), raising the possibility that the metastatic potential of slow-proliferating CRCs may be due to the presence of a high proportion of cancer stem cells.
Another possible reason for the association between low cellular proliferation and poor prognosis could be related to chronic and acute hypoxic regions present in CRCs. It has been shown that hypoxic conditions may slow down the growth rate of tumours but can promote the onset of an EMT leading to invasion (Yang et al, 2008).
Aneuploidy, an abnormal chromosome number, may provide an additional explanation for the association of low proliferation with high metastatic potential. Aneuploidy would be expected to have dramatic consequences on basic cellular functions such as proliferation. In budding yeast, for example, aneuploidy in the form of extra chromosome gains was found to result in a proliferative disadvantage (Torres et al, 2007). This same phenomenon occurs when mammalian cells carry additional chromosomes. In a recent publication, trisomic mouse embryonic fibroblasts were shown to proliferate more slowly than the euploid controls (Williams et al, 2008). Furthermore, a high level of chromosomal instability inhibits tumour growth, probably owing to inefficient cell proliferation or apoptotic induction (Weaver et al, 2007; Garcia-Higuera et al, 2008). All these studies suggest that aneuploidy hampers cell proliferation in human cells. These findings along with the established association between aneuploidy and a bad outcome in CRC (Araujo et al, 2007) suggest that the aneuploid state of more aggressive CRCs is one likely (but by no means the only) cause for the low proliferative activity of metastatic CRC. Possible underlying mechanisms are currently under investigation in our laboratory.
The extremely poor survival for patients with metastatic disease highlights the need for improved treatments. This requires the elucidation of the molecular and cellular mechanisms underlying CRC metastasis. The intriguing finding of an inverse relationship between tumour proliferation and metastasis suggests that slow proliferation is an important biological feature of a metastatic signature in CRC. This suggests that cytotoxic drugs killing fast-proliferating cells may not necessarily be the best choice for the treatment of metastatic disease. Therefore, the delineation of the mechanisms that underlie this biological feature of CRC may lead to the development of novel antimetastatic treatment strategies.
Conflict of interest
Anthony E Reeve is a director of Pacific Edge Biotechnology Ltd, Dunedin, New Zealand.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adam R, Vinet E (2004) Regional treatment of metastasis: surgery of colorectal liver metastases. Ann Oncol 15: iv103–iv106
Agui T, McConkey DJ, Tanigawa N (2002) Comparative study of various biological parameters, including expression of survivin, between primary and metastatic human colonic adenocarcinomas. Anticancer Res 22: 1769–1776
Anjomshoaa A, Lin YH, Black MA, McCall JL, Humar B, Song S, Fukuzawa R, Yoon HS, Holzmann B, Friederichs J, van Rij A, Thompson-Fawcett M, Reeve AE (2008) Reduced expression of a gene proliferation signature is associated with enhanced malignancy in colon cancer. Br J Cancer 99: 966–973
Araujo S, Bernardo W, Habr-Gama A, Kiss D, Cecconello I (2007) DNA ploidy status and prognosis in colorectal cancer: a meta-analysis of published data. Dis Colon Rectum 50: 1800–1810
Bird NC, Mangnall D, Majeed AW (2006) Biology of colorectal liver metastases: a review. J Surg Oncol 94: 68–80
Boman BM, Fields JZ, Cavanaugh KL, Guetter A, Runquist OA (2008) How dysregulated colonic crypt dynamics cause stem cell overpopulation and initiate colon cancer. Cancer Res 68: 3304–3313
Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T (2005) Migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat Rev Cancer 5: 744–749
Colantuoni C, Henry G, Zeger S, Pevsner J (2002) SNOMAD (Standardization and NOrmalization of MicroArray Data): web-accessible gene expression data analysis. Bioinformatics 18: 1540–1541
D’Arrigo A, Belluco C, Ambrosi A, Digito M, Esposito G, Bertola A, Fabris M, Nofrate V, Mammano E, Leon A, Nitti D, Lise M (2005) Metastatic transcriptional pattern revealed by gene expression profiling in primary colorectal carcinoma. Int J Cancer 115: 256–262
Dai H, van’t Veer L, Lamb J, He YD, Mao M, Fine BM, Bernards R, van de Vijver M, Deutsch P, Sachs A, Stoughton R, Friend S (2005) A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res 65: 4059–4066
Garcia-Higuera I, Manchado E, Dubus P, Canamero M, Mendez J, Moreno S, Malumbres M (2008) Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol 10: 802–811
Hara Y, Ogata Y, Shirouzu K (2000) Early tumor growth in metastatic organs influenced by the microenvironment is an important factor which provides organ specificity of colon cancer metastasis. J Exp Clin Cancer Res 19: 497–504
Jung A, Schrauder M, Oswald U, Knoll C, Sellberg P, Palmqvist R, Niedobitek G, Brabletz T, Kirchner T (2001) The invasion front of human colorectal adenocarcinomas shows co-localization of nuclear β-catenin, cyclin D1, and p16INK4A and is a region of low proliferation. Am J Pathol 159: 1613–1617
Kitabatake T, Kojima K, Fukasawa M, Beppu T, Futagawa S (2002) Correlation of thymidine phosphorylase staining and the Ki-67 labeling index to clinicopathologic factors and hepatic metastasis in patients with colorectal cancer. Surg Today 32: 322–328
Koehler A, Bataille F, Schmid C, Ruemmele P, Waldeck A, Blaszyk H, Hartmann A, Hofstaedter F, Dietmaier W (2004) Gene expression profiling of colorectal cancer and metastases divides tumours according to their clinicopathological stage. J Pathol 204: 65–74
Krasnoselsky AL, Whiteford CC, Wei JS, Bilke S, Westermann F, Chen Q-R, Khan J (2004) Altered expression of cell cycle genes distinguishes aggressive neuroblastoma. Oncogene 24: 1533–1541
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408
Palmqvist R, Sellberg P, Oberg A, Tavelin B, Rutegard JN, Stenling R (1999) Low tumour cell proliferation at the invasive margin is associated with a poor prognosis in Dukes’ stage B colorectal cancers. Br J Cancer 79: 577–581
Prall F (2007) Tumour budding in colorectal carcinoma. Histopathology 50: 151–162
Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, Gascoyne RD, Grogan TM, Muller-Hermelink HK, Smeland EB, Chiorazzi M, Giltnane JM, Hurt EM, Zhao H, Averett L, Henrickson S, Yang L, Powell J, Wilson WH, Jaffe ES, Simon R, Klausner RD, Montserrat E, Bosch F, Greiner TC, Weisenburger DD, Sanger WG, Dave BJ, Lynch JC, Vose J, Armitage JO, Fisher RI, Miller TP, LeBlanc M, Ott G, Kvaloy S, Holte H, Delabie J, Staudt LM (2003) The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 3: 185–197
Seong J, Cho JH, Yang WI, Chung EJ, Kim NK (2004) Apoptosis and proliferation in paired primary colorectal adenocarcinomas and their liver metastases. Yonsei Med J 45: 187–192
Steeg PS (2006) Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 12: 895–904
Thomas GV, Szigeti K, Murphy M, Draetta G, Pagano M, Loda M (1998) Down-regulation of p27 is associated with development of colorectal adenocarcinoma metastases. Am J Pathol 153: 681–687
Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924
Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW (2007) Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11: 25–36
Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A (2008) Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322: 703–709
Wolpin BM, Mayer RJ (2008) Systemic treatment of colorectal cancer. Gastroenterology 134: 1296–1310.e1
Yang M-H, Wu M-Z, Chiou S-H, Chen P-M, Chang S-Y, Liu C-J, Teng S-C, Wu K-J (2008) Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol 10: 295–305
Acknowledgements
We would like to express our gratitude to Pacific Edge Biotechnology Limited for supplying the microarray slides, and Dr Richard Simon and BRB-Array Tools Development Team. This research was funded by the Health Research Council of New Zealand and the Lottery Grants Board of New Zealand.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on British Journal of Cancer website (http://www.nature.com/bjc)
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Anjomshoaa, A., Nasri, S., Humar, B. et al. Slow proliferation as a biological feature of colorectal cancer metastasis. Br J Cancer 101, 822–828 (2009). https://doi.org/10.1038/sj.bjc.6605229
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605229
Keywords
This article is cited by
-
Nuclear control of lung cancer cells migration, invasion and bioenergetics by eukaryotic translation initiation factor 3F
Oncogene (2020)
-
A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer
Nature Communications (2020)
-
Frequent RNF43 mutation contributes to moderate activation of Wnt signaling in colorectal signet-ring cell carcinoma
Protein & Cell (2020)
-
Immunohistochemical investigation of prognostic biomarkers in resected colorectal liver metastases: a systematic review and meta-analysis
Cancer Cell International (2018)
-
Classification of gene signatures for their information value and functional redundancy
npj Systems Biology and Applications (2017)
