
Abstract
Dendritic cells (DCs) ‘pulsed’ with an appropriate antigen may elicit an antitumour immune response in mouse models. However, while attempting to develop a DC immunotherapy protocol for the treatment of breast cancer based on the tumour-associated MUC1 glycoforms, we found that unpulsed DCs can affect tumour growth. Protection from RMA-MUC1 tumour challenge was achieved in C57Bl/6 MUC1 transgenic mice by immunising with syngeneic DCs pulsed with a MUC1 peptide. However, unpulsed DCs gave a similar level of protection, making it impossible to evaluate the effect of immunisation of mice with DCs pulsed with the specific peptide. Balb/C mice could also be protected from tumour challenge by immunisation with unpulsed DCs prior to challenge with murine mammary tumour cells (410.4) or these cells transfected with MUC1 (E3). Protection was achieved with as few as three injections of 50 000 naïve DCs per mouse per week, was not dependent on injection route, and was not specific to cell lines expressing human MUC1. However, the use of Rag2-knockout mice demonstrated that the adaptive immune response was required for tumour rejection. Injection of unpulsed DCs into mice bearing the E3 tumour slowed tumour growth. In vitro, production of IFN-γ and IL-4 was increased in splenic cells isolated from mice immunised with DCs. Depleting CD4 T cells in vitro partially decreased cytokine production by splenocytes, but CD8 depletion had no effect. This paper shows that naïve syngeneic DCs may induce an antitumour immune response and has implications for DC immunotherapy preclinical and clinical trials.
Similar content being viewed by others

Main
The growth and metastatic spread of tumours depends in part on their ability to evade host immune surveillance and overcome host defences. All tumours express antigens that can be recognised by the immune system, but in most cases an inadequate immune response is elicited because of ineffective activation of effector cells by downregulation of major histocompatibility (MHC) molecules or inhibition of their function by factors produced by the tumour, such as TGF-β or IL-10 (Cheng et al, 2004; Houghton and Guevara-Patino, 2004; Campoli et al, 2005).
Immunotherapy using autologous dendritic cells (DCs) pulsed with tumour-specific antigens has been proposed as a therapeutic strategy for the treatment of a wide variety of tumours, and many preclinical and clinical trials of this approach using different antigens – including peptides and RNA – are underway (Meidenbauer et al, 2001; Schuler et al, 2003; Wierecky et al, 2006). Dendritic cells are potent antigen-presenting cells (APCs), which initiate and regulate innate and antigen-specific responses (Reis e Sousa, 2006). They originate from the bone marrow and their precursors home in via the bloodstream on almost all organs, where they are found in an immature state with high endocytic and phagocytic capability, continuously sampling the antigenic environment. Upon contact with an antigen, such as bacterial DNA, viral dsRNA, microbial products or with inflammatory cytokines, these interstitial DCs change their phenotype and function and migrate to the germinal centres of regional lymph nodes, where they present antigens to the resting or naïve T cells and induce antigen-specific T-cell responses.
MUC1, a highly glycosylated transmembrane glycoprotein, is overexpressed and aberrantly glycosylated by breast cancer cells resulting in changes in the antigenic profile of the tumour-associated glycoforms. In addition, T cells reactive with an HLA-A2*0201 class I epitope overlapping the MUC1 signal sequence (M1.2 epitope) have been demonstrated in breast cancer patients with this HLA type (Brossart et al, 1999; Beckhove et al, 2004; Correa et al, 2005). Mouse MHC class I epitopes particularly for H2b, as well as for other allotypes, have also been shown to be present in MUC1 (Apostolopoulos et al, 1998; Heukamp et al, 2001). Furthermore, it has been shown that lysate-pulsed DCs can cross-present the M1.2 epitope effectively and cross-prime CD8+ T cells in vitro (Bohnenkamp et al, 2004). Although it is not completely clear which components of the innate or adaptive immune system can be mobilised by different MUC1 immunogens and which are crucial for tumour rejection, evidence is accumulating that inhibition of tumour growth in mice and humans can be achieved. There are several reports presenting data from mouse models that suggest that loading DCs with MUC1-based immunogens can result in tumour rejection in MUC1 transgenic (MUC1.Tg) mice (Koido et al, 2000; Chen et al, 2003).
It has previously been reported that loading murine bone marrow-derived DCs with a MUC1 peptide and injecting them into mice protects the animals from subsequent challenge with MUC1-expressing breast tumours (Soares et al, 2001). In this paper, we present data that were obtained while attempting to pursue these observations and which show that protection from tumour challenge can be obtained using DCs that have not been loaded with any tumour-specific antigen. Using a different model system, a similar observation has been reported by the group of Berzofsky (van den Broeke et al, 2003). This phenomenon has important implications for the design of both pre-clinical and clinical DC-based protocols.
Materials and methods
Cell lines
Murine 410.4 (murine epithelial breast tumour cells derived from Balb/C mice) cells were a gift from Bonnie Miller (Michigan Cancer Foundation, Detroit, MI, USA) and were cultured in DMEM medium supplemented with 10% FCS and 25 μg ml−1 insulin. E3 cells, which express human MUC1 from its own promoter, were generated from 410.4 cells as described by Lalani et al (1991). RMA (murine leukaemia virus-induced T-cell lymphoma) cells were transfected with human MUC1 under the control of a cytomegalovirus (CMV) promoter and cultured as described previously (Plunkett et al, 2004).
Mouse strains
Female Balb/C and C57Bl/6 mice and transgenic mice (approximately 10 weeks of age) were used for bone marrow harvest and tumour challenge. MUC1.Tg mice expressing human MUC1 from the human MUC1 promoter, homozygous for the transgene expression, were originally developed on an H2-k background (Peat et al, 1992). These mice have been backcrossed onto C57Bl/6 and Balb/C strains for 15 generations to give a pure C57Bl/6 (H2b) or pure Balb/C (H2d) backgrounds. Recombination activation gene 2 (Rag2)–knockout (Rag2−/−) mice are on a C57Bl/6 background.
Generation of DCs
Bone marrow was isolated from the femurs and tibias of 8–12 weeks old female mice. Cells were resuspended in AIM-V medium in plastic culture dishes for 45 min; adherent cells were then discarded. Non-adherent and loosely adherent cells were resuspended in AIM-V medium supplemented with 50 μ M β-mercaptoethanol and 10 ng ml−1 IL-4 and GM-CSF (R&D Systems, Abingdon, UK). Fresh cytokines were added after 2 days culture. After 3 days of culture, non-adherent cells were removed and fresh medium supplemented with cytokines was added to the adherent cells. After a further 4 days, during which fresh cytokines were added every 48 h, non-adherent cells were collected and, where appropriate, primed with peptide. Dendritic cells were washed three times in endotoxin-free PBS prior to injections.
Flow cytometry
FITC- and PE-labelled mAbs against DC markers (BD Biosciences Pharmingen, San Jose, CA, USA) and isotype-matched labelled controls were used to characterise cell surface phenotypes by flow cytometry.
For staining, cells were washed and resuspended in PBS supplemented with 1% heat-inactivated FBS and 0.01% NaN3. Antibodies were diluted in this buffer and used at a final concentration of 25 μg ml−1. Incubations with antibodies were carried out for 45 min on ice. Following washing, labelled cells were fixed with 1% formaldehyde solution and 10 000 cells were analysed by flow cytometry.
Pulsing DCs
DCs were washed and resuspended in fresh medium with cytokines at a density of 106 cells per ml. Dendritic cells were ‘pulsed’ overnight with a 60-mer peptide consisting of three tandem repeats of MUC1 (20 μg ml−1). After pulsing, cells were washed three times in PBS.
Immunisation and tumour challenge
For protection experiments, mice received three subcutaneous injections (a week apart) of PBS containing (unless otherwise indicated) 5 × 104 DCs into the flank. A control group of mice were injected with PBS only. A week after the final DC injection, 0.1 ml PBS containing tumour cells (5 × 105 410.4 or E3 cells; 5 × 104 RMA-MUC1 cells) was injected subcutaneously into the flank of Balb/C (E3 and 410.4) or C57Bl/6 WT or transgenic mice (RMA-MUC1). For ethical reasons, mice were sacrificed when tumours reached 1.44 cm2. UKCCCR guidelines were followed at all times and all animal work was performed under Home Office Project licence No. PPL 70/4701.
Microchemotaxis assay
Chemotaxis was examined using a 48-well microchemotaxis chamber (Neuro Probe, Cabin John, MD, USA). The lower wells were filled with RPMI supplemented with 250 ng ml−1 chemokine and covered with an 8-μ M-pore polycarbonate membrane. Cell suspension (100 000 DCs in 100 μl serum-free medium) was added to each upper well. After incubation for 2 h at 37°C for 90 min, the membrane was removed and the cells attached to the upper surface of the membrane were removed by washing with PBS. The membrane was fixed in methanol and stained with Diff-Quik. Cells attached to the lower surface of the membrane or in the lower well of chamber were counted.
Splenocyte preparation
Spleens were disaggregated and the cells suspended in RPMI medium before centrifugation at 1500 rpm for 10 min. Splenocytes were isolated using a Ficoll–Paque gradient (GE Healthcare UK Ltd, Buckinghamshire, UK), and cell clumps were removed by filtering the cells through 30 μ M filters. Cells were washed twice with RPMI and resuspended (107 cells per ml) in RPMI supplemented with 5% FCS and 10 μ M β-mercaptoethanol, before being plated in a 96-well culture plate (100 μl per well) with or without 106 unpulsed DCs. CD4- or CD8-positive cells were removed in vitro prior to culture using commercially available BD IMag Particles (BD Biosciences Pharmingen) according to the manufacturer's instructions.
Cytokine determination
Cytokine (IFN-γ and IL-4) concentrations in cell culture supernatants were measured by ELISAs from R&D Systems according to the manufacturer's instructions.
Results
Phenotypic and functional analysis of murine DCs
DCs were cultured from the bone marrow of wild-type or MUC1. Tg mice on Balb/c and C57Bl/6 backgrounds in serum-free medium. The phenotype of the cells was analysed by flow cytometry: cells from all strains of mice were CD11b+ve, CD11c+ve, CD80+ve, CD86+ve, MHC class II+ve, CXCR4weakly +ve and CD8a−ve. Figure 1A shows the phenotype of DCs derived from C57Bl/6 MUC1.Tg mice. Dendritic cells derived from the bone marrow of wild-type C57Bl/6 and Balb/c mice had a similar phenotype (data not shown). The DCs used in this study were therefore phenotypically mature and functionally active as shown by their ability to migrate towards chemokines (Figure 1B) and to take up FITC-dextran (Figure 1C). Pulsing cells with MUC1 peptide did not alter DC phenotype (data not shown). Stimulating cells with either LPS or prostaglandin E2/tumour necrosis factor α increased cell surface CXCR4 but did not increase in vitro migration (data not shown).

Bone marrow-derived DCs grown in serum-free medium show a typical dendritic phenotype and are functionally active. (A) Bone marrow-derived DCs from C57Bl/6 MUC1.Tg mice were cultured for 6 days in serum-free medium and their phenotype determined by flow cytometry. (B) Migration towards chemokines of 6-day cultured DCs was measured in the transwell system. (C) The ability of DCs to uptake FITC dextran was measured at 4°C (thick line) and at 37°C (shaded).
Vaccination with pulsed or unpulsed DCs can protect mice from tumour challenge
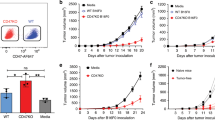
To investigate the ability of DCs pulsed with MUC1 peptides to protect mice from challenge with MUC1-expressing tumours, we vaccinated C57Bl/6 MUC1.Tg. mice with MUC1 peptide-pulsed DCs, similar to the experiments described by Soares et al (2001). C57Bl/6 MUC1.Tg mice were vaccinated with three injections, 3 weeks apart, of syngeneic MUC1.Tg DCs, pulsed or unpulsed with MUC1 peptide. One week after the last injection, the mice were challenged with RMA tumour cells expressing human MUC1 under a CMV promoter. Although peptide-pulsed DCs significantly increased the survival of the mice compared with the buffer control group, unpulsed DCs also gave significant protection so that by day 56, the protection from naïve DCs was indistinguishable from that obtained with the pulsed DCs (Figure 2A). This made it impossible to evaluate the effect of immunisation of mice with DCs pulsed with the specific peptide.

Naïve DCs as well as DCs pulsed with antigen can protect mice from tumour challenge. (A) Groups of seven C57Bl/6 MUC1.Tg mice were vaccinated with three injections of syngeneic DCs given 3 weeks apart either pulsed with a 60-mer peptide corresponding to three tandem repeats of MUC1 or naïve unpulsed DCs. Control mice were vaccinated with PBS. At 1 week after the third injection, the mice were challenged with RMA tumour cells expressing human MUC1. For ethical reasons the mice were sacrificed when the tumour reached 1.4 cm2. (B) Wild-type Balb/C mice (five per group) were given three weekly subcutaneous injections of either PBS or 5 × 105 unpulsed DCs. A week after the final DC injection, mice were challenged with subcutaneous injection of 500 000 E3 breast tumour cells. Shown is one representative experiment of three.
To ascertain whether the protection from tumour challenge provided by unpulsed DCs was limited to this model, we changed the mouse strain and the tumour cell line. Wild-type Balb/C mice were treated with three weekly subcutaneous injections of unpulsed wild-type DCs (50 000 cells per mouse per week). A week after the final DC injection, the mice were challenged with E3 tumour cells (the murine mammary tumour cell line 410.4 transfected with human MUC1; Figure 2B). In the PBS control groups, all mice had a tumour within 3 weeks of tumour challenge. However, unpulsed DCs significantly increased the tumour-free survival of the mice, with 60% of the mice remaining tumour-free until the termination of the experiment at 140 days. Thus, in two different murine models, immunisation with naïve DCs can protect mice from tumour challenge.
Protection is dependent on the number of injected DCs
We analysed the effects of different numbers of DCs on tumour protection in two different mouse strains. Wild-type Balb/C mice were vaccinated with zero to 500 000 DCs per week and after three DC injections, mice were challenged with the E3 tumour. Injections of 5000 DCs per mouse per week did not protect the mice from subsequent tumour challenge (Figure 3A). However, injection of 50 000 or 500 000 resulted in a significant number of mice remaining tumour-free up until the termination of the experiment at day 140. Increasing the number of DCs above 50 000 did not further improve tumour protection.

Protection from tumour challenge is dependent on the number of DCs injected but not on the route of injection. (A) Balb/C mice were injected three times with 0, 5 × 103, 5 × 104 or 5 × 105 DCs per mouse per week prior to tumour challenge with E3 cells. (B) Three weekly injections of 5 × 104 DCs were administered to wild-type Balb/c mice subcutaneously in the right or left flank or intravenously into the tail vein. At 1 week after the final injection, the mice were tumour challenged with E3 cells.
Protection is not compromised by route of injection
In the previous experiments, immunisation injections were at the same site as tumour challenge (ie, subcutaneous injections in the left flank). We next tested whether the tumour challenge in the same site as DC injection effectively ‘pulsed’ the DCs in vivo, or whether an inflammatory effect at the tumour challenge site was ‘activating’ DCs, by varying the route of injection. Balb/C mice were given three injections of unpulsed DCs or PBS into either the right or left flank; DCs were also injected intravenously. The subsequent E3 tumour challenge was injected into the left flank of all groups. Injection of unpulsed DCs gave a similar level of tumour protection whether the injection site was left flank or intravenous (Figure 3B), indicating that tumour protection does not derive from the synergy of the DCs and an inflammatory response or from ‘in vivo pulsing’.
Protection from tumour challenge is not dependent on MUC1 but does require T or B cells
The E3 murine mammary tumour cell line expresses human MUC1. To test whether DC-mediated protection from tumour challenge depended on the presence of a foreign antigen in the tumour cells (ie, human MUC1), we tumour challenged naïve DC immunised mice with E3 cells or parental wild-type 410.4 cells. Wild-type Balb/C mice were immunised with unpulsed DCs derived from wild-type Balb/C bone marrow prior to tumour challenge. As previously observed, naïve DCs protected Balb/C mice from E3 tumour challenge. However, mice were also protected from challenge with the syngeneic parental cell line, 410.4 (Figure 4A).

Protection from tumour challenge is not dependent on MUC1 expression but does require B or T cells. (A) Wild-type Balb/C mice (eight per group) were given three weekly subcutaneous injections of either PBS or 5 × 105 unpulsed DCs. A week after the final DC injection, mice were challenged with subcutaneous injection of either 410.4 or E3 cells (410.4 transfected with human MUC1) (500 000 tumour cells per mouse). (B) C57Bl6 and Rag2−/− mice were vaccinated with 50 000 DCs (three times, 1 week apart) before challenge with the RMA tumour cell line.
To investigate the mechanism involved in the tumour protection, we immunised Rag2−/− mice with naïve DC and tumour challenged with RMA cells. The phenotype of Rag2−/− mice, which are on a C57Bl6 background, is mature T- and B-cell deficiency. Although the Rag2−/− mice developed tumours more quickly than the wild-type C57Bl6 mice, by day 13 all the Rag2−/−mice in the control group and the naïve DC group had developed tumours and there was no difference in the tumour growth between the two groups (Figure 4B). In contrast, in the wild-type mice, vaccination with naïve DCs reduced the take and growth of the RMA tumour cells compare with the wild-type controls (Figure 4B).
These data suggest that either T or B cells are involved in the tumour protection observed with naïve DC cells and that the protection is not dependent on the presence of a foreign antigen.
Naïve DCs induce the production of IFN-γ and IL-4 by splenocytes, which is dependent on T-cell function
To investigate the immune response induced by naïve DCs, wild-type C57Bl/6 mice were immunised three times with naïve DCs; 1 week after the final injection, the spleens were removed and the splenocytes cultured for 48 h in the presence or absence of syngeneic DCs. Incubation of naïve DCs with splenocytes from mice vaccinated with unpulsed DCs resulted in the secretion of high levels of IFN-γ and IL-4 (Figure 5A and B). No IFN-γ or IL-4 was detected when DCs were incubated with splenocytes from mice injected with PBS. Thus the vaccination of mice with DCs cultured for 7 days in vitro, without exposure to specific antigen, was sufficient to induce the in vitro secretion of IFN-γ and IL-4. It should be noted that the DCs used to immunise the mice were cultured in serum-free medium, thus eliminating any artefacts due to the foetal calf sera.

Unpulsed DCs can stimulate in vivo primed splenocytes to secrete cytokines in vitro. Mice were injected three times with either PBS or naïve DCs; 1 week after the last injection, the splenocytes were isolated and cultured for 48 h in the presence or absence of naïve DCs and the cytokines produced measured by ELISA. (A) IFN-γ production by splenocytes from mice immunised with DCs or PBSA cultured in vitro in the presence (clear bars) or absence (black bars) of naïve DCs. Measurements from eight individual splenocyte preparations are shown; (B) IL-4 production by splenocytes from mice immunised with DCs or PBSA cultured in vitro in the presence (clear bars) or absence (black bars) of naïve DCs. Measurements from eight individual splenocyte preparations are shown. (C and D) CD4+ cells or CD8+ cells were depleted from splenocytes of mice vaccinated with naïve DCs prior to in vitro culture in the presence or absence (negative control) of naïve DCs. (C) IFNγ secretion (*, P<0.05), the mean of four splenocyte preparations±s.d. is shown. (D) IL-4 secretion (*, P<0.05), the mean of four splenocyte preparations±s.d. is shown.
To determine the source of the cell producing the IFN-γ and IL-4, the CD4+ or CD8+ T cells were depleted prior to the incubation with DCs. The efficacy of depletion was analysed by flow cytometry and shown to be 93.0±0.5% for CD4+ve and 68.4±10.4% for CD8+ve T cells (data not shown). Depleting the splenocytes of CD4 cells partially but significantly decreased the induction of IFN-γ (Figure 5C) and IL-4 (Figure 5D) release, whereas depleting CD8 cells had no effect.
Therapy of E3 tumour with unpulsed DCs
Although protection from tumour challenge has been attained using immunotherapy and vaccination methods, therapy of established tumours is usually less effective. Therefore, having shown induction of protection against the E3 tumour using unpulsed DCs, we tested whether naïve DCs had any effect on the more stringent system of the established tumour. Balb/C mice were challenged with E3 tumour cells and then given three weekly subcutaneous injections of naïve DCs or PBS.
Immunisation with DCs did not prevent tumour growth (Figure 6A), but tumour growth was significantly less in mice immunised with naïve DCs (Figure 6B and C).

Therapy of E3 tumour-bearing mice by immunisation with naïve DC. Groups of eight Balb/C mice were subcutaneously injected with E3 cells. After 7 days, weekly subcutaneous injections of PBS, unpulsed DCs (105 cells per mouse per week) began, for 3 weeks. (A) Tumour-free survival of the mice, (B) growth of tumours over time. (C) Comparison of the average size of the tumours at day 21. The tumour area was significantly (*, P<0.05) smaller in the DC-treated group compared with PBS-treated mice.
Discussion
Immunotherapy using autologous DCs pulsed with a tumour-specific antigen has been proposed as a therapeutic strategy for the treatment of a wide variety of tumours, and many preclinical and clinical trials of this approach are underway (Meidenbauer et al, 2001). Indeed, several studies have shown an encouraging clinical response in both clinical trials and numerous animal models (Schuler et al, 2003; Wierecky et al, 2006). One of the critical factors shown to influence the efficacy of DC immunotherapy has been the preparation and differentiation of DCs (Josien et al, 2000; Liu et al, 2002; Schuler et al, 2003). In this study, we have cultured murine bone marrow cells in the presence of IL-4 and GM-CSF to yield high-quality CD11b+ve CD11c+ve CD8−ve myeloid mature DCs that express high levels of stimulatory MHC class II molecules, as well as CD80, CD86 and the chemokine receptor CXCR4. Furthermore, it was demonstrated that the generated DCs are functionally active by migrating effectively towards CCL19 and CXCL12, the ligands for CCL7 and CXCR4, respectively. We show that injection of mice with DCs pulsed with a MUC1 tandem repeat peptide (60-mer corresponding to three tandem repeats) can protect wild-type and MUC1.Tg mice from subsequent challenge with a MUC1-expressing tumour. However, we also demonstrated that injection of unpulsed ‘naïve’ DCs are equally efficient at protecting mice from tumour challenge. We have shown that the protection gained by injection of naïve DCs in our studies is not due to (1) the expression of a foreign (human) antigen, (2) a high number of DCs being injected, (3) the route of immunisation, suggesting the induction of systemic protection, (4) strain of mouse, or (5) tumour model. Using Rag2−/− mice we have demonstrated that the protection observed with naïve DCs was dependent on elements of the adaptive immune response, that is T or B cells.
Moreover, in vivo priming with syngeneic unpulsed DCs resulted in the secretion of large amounts of IFN-γ and IL-4 upon a single in vitro stimulation with naïve DCs.
Interestingly, it has been shown that DCs isolated from mouse spleens spontaneously produce IL-12 and TNF-α, upregulate costimulatory molecules and induce the activation of antigen-specific IFN-γ-producing CD4+ T cells in vivo (Schlecht et al, 2006). The responses were similar to those induced by DCs activated by CpG, a strong Th1-promoting adjuvant (Klinman, 2006). Moreover, DCs derived in vitro from bone marrow have been shown to secrete type I IFNs that act in an autocrine manner to activate the DCs enabling them to activate T cells (Montoya et al, 2002). The DCs used in this study express relatively high levels of CD80, CD86 and class II and possibly were more activated than those used in other studies. Dendritic cells can induce tolerance or immunity according to their activation state (Moser, 2003; Reis e Sousa, 2006); it is possible that DCs activated in vitro by the culture conditions could present self-antigens – common to the tumour cells – to T cells resulting in their activation. Certainly, effector T cells have been shown to respond to a lower ligand affinity threshold than naïve T cells, and effector T cells have been shown to respond to endogenous self-peptide presented by APCs (Kimachi et al, 2003). Further experiments will be needed to investigate whether there is evidence of induced autoimmunity in our models.
To minimise the exposure of the DCs to foreign proteins, the DCs were cultured in serum-free AIM V medium. However, it cannot be ruled out that the DCs presented proteins found within the medium to T cells in vivo and that the in vitro cytokine production observed with unpulsed DCs resulted from a stimulation of these cells by the DCs. This could be possible for although the splenocytes were incubated with DCs in a completely different medium (RPMI 10% FCS), the DCs used for the in vitro assay were derived from bone marrow in AIM-V medium. However, this cannot explain the ability of naïve DCs to protect mice from tumour challenge, and other groups have used AIM-V to generate bone marrow-derived DCs (Soares et al, 2001).
There are many reports in the literature of the use of primed DCs to induce tumour protection in murine models. Most of these (Zitvogel et al, 1996; Ashley et al, 1997; Boczkowski et al, 2000; Irvine et al, 2000; Nair et al, 2000; Shimizu et al, 2001; Koido et al, 2002; Chen et al, 2003) have included control DC groups, whereas others have not (Soares et al, 2001). In reports that included a control group of unpulsed DCs, no significant protection was seen with these cells. However, in agreement with our data, the study by van den Broeke et al (2003) showed that unpulsed DCs induced protection against tumour lung metastases. As we have found, this protection was independent of the strain of mouse, tumour source or route of injection of the DCs. Moreover, they showed that CD4+ T cells were necessary for protection together with NK cells. The authors conclude that the DC-mediated NK cell activation was likely to be through an intermediate interaction of DCs with CD4+ T cells rather than a direction effect on the NK cells. Furthermore, although small in number there have been other reports that unpulsed DCs can induce tumour protection (Yang et al, 1997; DeMatos et al, 1998). Interestingly, early studies by Knight (Knight et al, 1985) also suggested that normal syngeneic DCs could induce tumour regression or delayed tumour growth.
From the results presented here it is clear that manipulation of DCs in vitro can result in their ability to stimulate an immune response without actively pulsing the DCs with antigen. One of the hurdles in cancer immunotherapy has been to show a connection between any clinical benefit and the antigen with which the DCs were pulsed. In a stage IV melanoma clinical phase I study using four melanoma antigens with BM-derived DCs, none of the patients analysed showed an expansion of melanoma-peptide-specific circulating effector memory T cells, and there were no objective clinical responses (Banchereau et al, 2005). However, in other studies, there were detectable levels of CD4-specific Th1 cells and specific CD8 T cells in most but not all of the patients (Schultz et al, 2004; Fay et al, 2006).
The fact that unpulsed DCs can induce T cells to produce large amounts of cytokines (IFN-γ and IL-4) and protect mice from tumour challenge has implications for the use of DCs in immunotherapy. Understandably, unpulsed DCs have not been included in the early clinical trials investigating the efficacy of DCs for the treatment of cancer patients. It is possible that some of the promising results reported are not dependent on the antigen used to pulse the DC ex vivo. More importantly, if injection of naïve ‘culture-activated’ DCs could induce T cells to self-antigens, there is a possibility of inducing autoimmunity in patients undergoing such therapy and therefore further research into the results reported here is required.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Apostolopoulos V, Lofthouse SA, Popovski V, Chelvanayagam G, Sandrin MS, McKenzie IF (1998) Peptide mimics of a tumor antigen induce functional cytotoxic T cells. Nat Biotechnol 16: 276–280
Ashley DM, Faiola B, Nair S, Hale LP, Bigner DD, Gilboa E (1997) Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med 186: 1177–1182
Banchereau J, Ueno H, Dhodapkar M, Connolly J, Finholt JP, Klechevsky E, Blanck JP, Johnston DA, Palucka AK, Fay J (2005) Immune and clinical outcomes in patients with stage IV melanoma vaccinated with peptide-pulsed dendritic cells derived from CD34+ progenitors and activated with type I interferon. J Immunother 28: 505–516
Beckhove P, Feuerer M, Dolenc M, Schuetz F, Choi C, Sommerfeldt N, Schwendemann J, Ehlert K, Altevogt P, Bastert G, Schirrmacher V, Umansky V (2004) Specifically activated memory T cell subsets from cancer patients recognize and reject xenotransplanted autologous tumors. J Clin Invest 114: 67–76
Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E (2000) Induction of tumor immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Res 60: 1028–1034
Bohnenkamp HR, Coleman J, Burchell JM, Taylor-Papadimitriou J, Noll T (2004) Breast carcinoma cell lysate-pulsed dendritic cells cross-prime MUC1-specific CD8+ T cells identified by peptide-MHC-class-I tetramers. Cell Immunol 231: 112–125
Brossart P, Heinrich KS, Stuhler G, Behnke L, Reichardt VL, Stevanovic S, Muhm A, Rammensee HG, Kanz L, Brugger W (1999) Identification of HLA-A2-restricted T-cell epitopes derived from the MUC1 tumor antigen for broadly applicable vaccine therapies. Blood 93: 4309–4317
Campoli M, Ferrone S, Zea AH, Rodriguez PC, Ochoa AC (2005) Mechanisms of tumor evasion. Cancer Treat Res 123: 61–88
Chen D, Xia J, Tanaka Y, Chen H, Koido S, Wernet O, Mukherjee P, Gendler SJ, Kufe D, Gong J (2003) Immunotherapy of spontaneous mammary carcinoma with fusions of dendritic cells and mucin 1-positive carcinoma cells. Immunology 109: 300–307
Cheng F, Gabrilovich D, Sotomayor EM (2004) Immune tolerance in breast cancer. Breast Dis 20: 93–103
Correa I, Plunkett T, Coleman J, Galani E, Windmill E, Burchell JM, Taylor-Papdimitriou J (2005) Responses of human T cells to peptides flanking the tandem repeat and overlapping the signal sequence of MUC1. Int J Cancer 115: 760–768
DeMatos P, Abdel-Wahab Z, Vervaert C, Seigler HF (1998) Vaccination with dendritic cells inhibits the growth of hepatic metastases in B6 mice. Cell Immunol 185: 65–74
Fay JW, Palucka AK, Paczesny S, Dhodapkar M, Johnston DA, Burkeholder S, Ueno H, Banchereau J (2006) Long-term outcomes in patients with metastatic melanoma vaccinated with melanoma peptide-pulsed CD34(+) progenitor-derived dendritic cells. Cancer Immunol Immunother 55: 1209–1218
Heukamp LC, van der Burg SH, Drijfhout JW, Melief CJ, Taylor-Papadimitriou J, Offringa R (2001) Identification of three non-VNTR MUC1-derived HLA-A*0201-restricted T-cell epitopes that induce protective anti-tumor immunity in HLA-A2/K(b)-transgenic mice. Int J Cancer 91: 385–392
Houghton AN, Guevara-Patino JA (2004) Immune recognition of self in immunity against cancer. J Clin Invest 114: 468–471
Irvine AS, Trinder PK, Laughton DL, Ketteringham H, McDermott RH, Reid SC, Haines AM, Amir A, Husain R, Doshi R, Young LS, Mountain A (2000) Efficient nonviral transfection of dendritic cells and their use for in vivo immunization. Nat Biotechnol 18: 1273–1278
Josien R, Li HL, Ingulli E, Sarma S, Wong BR, Vologodskaia M, Steinman RM, Choi Y (2000) TRANCE, a tumor necrosis factor family member, enhances the longevity and adjuvant properties of dendritic cells in vivo. J Exp Med 191: 495–502
Kimachi K, Sugie K, Grey HM (2003) Effector T cells have a lower ligand affinity threshold for activation than naive T cells. Int Immunol 15: 885–892
Klinman DM (2006) Adjuvant activity of CpG oligodeoxynucleotides. Int Rev Immunol 25: 135–154
Knight SC, Hunt R, Dore C, Medawar PB (1985) Influence of dendritic cells on tumor growth. Proc Natl Acad Sci USA 82: 4495–4497
Koido S, Kashiwaba M, Chen D, Gendler S, Kufe D, Gong J (2000) Induction of antitumor immunity by vaccination of dendritic cells transfected with MUC1 RNA. J Immunol 165: 5713–5719
Koido S, Tanaka Y, Chen D, Kufe D, Gong J (2002) The kinetics of in vivo priming of CD4 and CD8 T cells by dendritic/tumor fusion cells in MUC1-transgenic mice. J Immunol 168: 2111–2117
Lalani EN, Berdichevsky F, Boshell M, Shearer M, Wilson D, Stauss H, Gendler SJ, Taylor-Papadimitriou J (1991) Expression of the gene coding for a human mucin in mouse mammary tumor cells can affect their tumorigenicity. J Biol Chem 266: 15420–15426
Liu A, Takahashi M, Narita M, Zheng Z, Kanazawa N, Abe T, Nikkuni K, Furukawa T, Toba K, Fuse I, Aizawa Y (2002) Generation of functional and mature dendritic cells from cord blood and bone marrow CD34+ cells by two-step culture combined with calcium ionophore treatment. J Immunol Methods 261: 49–63
Meidenbauer N, Andreesen R, Mackensen A (2001) Dendritic cells for specific cancer immunotherapy. Biol Chem 382: 507–520
Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, Tough DF (2002) Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 99: 3263–3271
Moser M (2003) Dendritic cells in immunity and tolerance-do they display opposite functions? Immunity 19: 5–8
Nair SK, Heiser A, Boczkowski D, Majumdar A, Naoe M, Lebkowski JS, Vieweg J, Gilboa E (2000) Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med 6: 1011–1017
Peat N, Gendler SJ, Lalani N, Duhig T, Taylor-Papadimitriou J (1992) Tissue-specific expression of a human polymorphic epithelial mucin (MUC1) in transgenic mice. Cancer Res 52: 1954–1960
Plunkett T, Graham R, Correa I, Sewell R, Miles D, Burchell J, Taylor-Papadimitriou J (2004) Protection against MUC1 expressing mouse tumours by intra-muscular injection of MUC1 cDNA requires functional CD8+ and CD4+ T cells but does not require the MUC1 tandem repeat domain. Int J Cancer 109: 691–697
Reis e Sousa C (2006) Dendritic cells in a mature age. Nat Rev Immunol 6: 476–483
Schlecht G, Mouries J, Poitrasson-Riviere M, Leclerc C, Dadaglio G (2006) Purification of splenic dendritic cells induces maturation and capacity to stimulate Th1 response in vivo. Int Immunol 18: 445–452
Schuler G, Schuler-Thurner B, Steinman RM (2003) The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol 15: 138–147
Schultz ES, Schuler-Thurner B, Stroobant V, Jenne L, Berger TG, Thielemanns K, van der Bruggen P, Schuler G (2004) Functional analysis of tumor-specific Th cell responses detected in melanoma patients after dendritic cell-based immunotherapy. J Immunol 172: 1304–1310
Shimizu K, Thomas EK, Giedlin M, Mule JJ (2001) Enhancement of tumor lysate- and peptide-pulsed dendritic cell-based vaccines by the addition of foreign helper protein. Cancer Res 61: 2618–2624
Soares MM, Mehta V, Finn OJ (2001) Three different vaccines based on the 140-amino acid MUC1 peptide with seven tandemly repeated tumor-specific epitopes elicit distinct immune effector mechanisms in wild-type vs MUC1-transgenic mice with different potential for tumor rejection. J Immunol 166: 6555–6563
van den Broeke LT, Daschbach E, Thomas EK, Andringa G, Berzofsky JA (2003) Dendritic cell-induced activation of adaptive and innate antitumor immunity. J Immunol 171: 5842–5852
Wierecky J, Mueller M, Brossart P (2006) Dendritic cell-based cancer immunotherapy targeting MUC-1. Cancer Immunol Immunother 55: 63–67
Yang S, Darrow TL, Vervaert CE, Seigler HF (1997) Immunotherapeutic potential of tumor antigen-pulsed and unpulsed dendritic cells generated from murine bone marrow. Cell Immunol 179: 84–95
Zitvogel L, Mayordomo JI, Tjandrawan T, DeLeo AB, Clarke MR, Lotze MT, Storkus WJ (1996) Therapy of murine tumors with tumor peptide-pulsed dendritic cells: dependence on T cells, B7 costimulation, and T helper cell 1-associated cytokines. J Exp Med 183: 87–97
Acknowledgements
This work was supported by Cancer Research UK and a grant from the European Commission, contract number QLK3-CT-2002-01980. We thank Gary Martin and Del Watling of CR-UK Biological Services, Clare Hall for their excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Grimshaw, M., Papazisis, K., Picco, G. et al. Immunisation with ‘naïve’ syngeneic dendritic cells protects mice from tumour challenge. Br J Cancer 98, 784–791 (2008). https://doi.org/10.1038/sj.bjc.6604221
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6604221
