
Abstract
There is no consensus as to the management of untreated poor prognosis or relapsed/refractory germ cell tumours. We have studied an intensive cisplatin-based regimen that incorporates high-dose methotrexate (HD MTX) and actinomycin-D and etoposide every 14 days (GAMEC). Sixty-two patients were enrolled in a phase 2 study including 27 who were untreated (IGCCCG, poor prognosis) and 35 with progression despite conventional platinum based chemotherapy. The pharmacokinetics of the drugs were correlated with standard outcome measures. Twenty of the untreated patients were progression free following GAMEC and appropriate surgery, as were 18 individuals in the pretreated group. None of the established prognostic factors for therapy for pretreated patients could identify a poor-prognosis group. Five out of nine late relapses to prior chemotherapy were progression free following GAMEC and appropriate surgery. All patients had at least one episode of febrile neutropenia and there were five (8%) treatment-related deaths. PK values were not predictive of efficacy or toxicity, although the dose intensity in the pretreated group of patients, especially of HD MTX, was significantly correlated with progression-free survival (PFS). GAMEC is a novel intensive regimen for this group of patients producing encouraging responses, although with significant toxicity. For those in whom it fails, further therapy is still possible with durable responses being seen.
Similar content being viewed by others

Main
The treatment of germ cell tumours (GCT) is one of the major successes of cytotoxic chemotherapy, with the three-drug BEP regimen (bleomycin, etoposide and cisplatin) being most established. Four cycles of BEP are considered optimal for those with untreated metastatic disease with the poorest prognosis in the routinely used IGCCCG classification (International Germ Cell Collaborative Group, 1997). For these individuals, the cure rate is approximately 50%, and to date, no regimen in any randomised controlled trial has shown a superior outcome (Kaye et al, 1998; Hinton et al, 2003), although higher cure rates have been seen in various phase II studies (Horwich et al, 2006).
Patients who relapse after first-line therapy have a durable cure rate of between 25–60%, with the most significant prognostic factors being the site of the primary, the response to initial therapy, the duration of this response and the level of serum tumour marker at relapse (McCaffrey et al, 1997). Cisplatin, ifosfamide and a third drug (paclitaxel, etoposide or vinblastine) are used most commonly with or without high-dose chemotherapy consolidation (Rick et al, 2001; Kondagunta et al, 2005; Mead et al, 2005). Others have used a tandem or triple stem cell transplant after initial stem cell mobilisation (Bhatia et al, 2000; Motzer et al, 2000).
On the basis of encouraging phase 2 data, several more complex regimens have been utilised for untreated patients, and these have generally intensified the drug cisplatin using a dose-dense approach (including BOP/BEP (Anthoney et al, 2004), CBOP/BEP (Christian et al, 2003)) or an approach in which additional agents are added (such as POMB/ACE; Bower et al, 1997). In certain patients, an etoposide dose of 500 mg m−2 is superior to 360 mg m−2 (Toner et al, 2001) (its absence from the initial few weeks of the BOP/VIP-B regimen may account for the failure to show an advantage of that treatment over BEP (bleomycin, etoposide and cisplatin); Kaye et al, 1998).
In relapsed disease, we have found that a dose-dense cisplatin using m-BOP (weekly cisplatin, MTX, bleomycin and vincristine), led to long-term remissions in 42% of patients (Shamash et al, 1999). In addition, the combination of etoposide, actinomycin-D and MTX (EAM) without cisplatin produced a complete remission (CR) rate of 21% in patients who had relapsed after PVB (Levi et al, 1990). This led us to develop granulocyte colony stimulating factor, actinomycin-D, methotrexate, etoposide, cisplatin (GAMEC), which was designed to achieve dose-dense delivery of cisplatin, early introduction of etoposide in a similar dose to conventional BEP, the incorporation of sliding scale MTX at high dose and add actinomycin-D to the therapy, with appropriate colony factor support.
Pharmacokinetics (PK) relationships for toxicity and efficacy endpoints have been documented for high-dose methotrexate (HD MTX) in osteosarcoma (Crews et al, 2004) and acute lymphoblastic leukaemia (ALL), and for teniposide in ALL (Evans et al, 1998). As this is a novel regimen, PK studies on MTX, 7-hydroxy MTX (its metabolite implicated in renal dysfunction), etoposide and actinomycin-D were conducted to identify possible relationships between PK parameters and either toxicity or efficacy. We also investigated the overall survival, time to progression and toxicities using this regimen.
Patients and methods
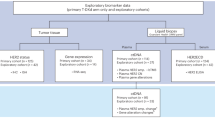
St Bartholomew's Hospital, London is one of the largest referral centres for GCT in the United Kingdom. Between September 1997 and June 2005, 62 patients were recruited for a phase 2 trial of GAMEC chemotherapy, with appropriate ethical approval and written informed consent. Twenty-seven were untreated patients with poor prognosis disease using the IGCCCG criteria and 35 had relapsed following at least one line of conventional platinum-based therapy.
Relapse was defined as the appearance of new disease or the development of increasing tumour markers in patients with known sites of disease. Patients receiving first-line therapy whose markers although not declining at the rate predicted by their known half-lives, were nevertheless still falling, were ineligible. Such patients completed their current therapy and only became eligible if their markers started to rise. Histological diagnosis was not mandatory in untreated patients who had a testicular mass and elevated tumour markers, if it were felt that delay of chemotherapy would compromise survival; all patients were staged using whole-body CT before therapy and all had renal function assessed using EDTA glomerular filtration rate (GFR) clearances. The GAMEC schedule is shown in Figure 1.

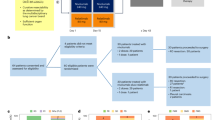
GAMEC chemotherapy schedule. Week 1: actinomycin-D (1 mg m−2) day 1: etoposide: 90 mg m−2, days 1–4 over 2 h; MTX loading dose over 30 min followed by 12 h infusion; day starting 4 h after the etoposide: cisplatin 50 mg m−2 over 4 h days 3 and 4. MTX was given as follows, depending on GFR: >120 ml min−1, 2 g m−2 loading then 8 g m−2 over 12 h (use 6g m−2 over 12 h if >30 years or PS>1); 100–119 ml min−1, 2 g m−2 loading, then 6 g m−2 over 12 h; 80–99 ml min−1, 2 g m−2 loading, then 4 g m−2 over 12 h; 60–79 ml min−1, 2 g m−2 loading, then 3 g m−2 over 12 h; 40–59 ml min−1, 1.5 g m−2 loading followed by 2 g m−2 over 12 h; 20–39 ml min−1, 1 g m−2 loading only. Acetazolamide was prescribed 500 mg 2 × per day for 3 days. Regular sodium bicarbonate (100 mmol), 6 h with 20 mmol KCl in 5% glucose for 48 h. Folinic acid was started 30 h post-MTX treatment. Folinic acid: rescue commenced at 30 h post-MTX treatment. The first level of MTX was taken 24 h post-infusion start. Initial rescue was 100 mg i.v. over 30 min, followed by 250 mg over 24 h. If the 24-h level was <2 μmol l−1 continue with folinic acid 30 mg, six hourly for 3 days, otherwise give a further infusion of 350 mg over 24 h and recheck level at 48 h. If the 24 h level >40μmol l−1 the folinic acid was increased to 700 mg per 24 h. At 48 h and beyond: if the level were >3 μ l−1 in the presence of renal impairment, the use of carboxypeptidase was considered. If renal function was preserved, an infusion of 700 mg per 24 h was continued; if the level was >1 and <3μmol l−1, 350 mg was given over 24 h, and the level was checked every 24 h. When the level was <1, the dose was reduced to 30 mg orally 4 × per day, until the level was <0.2 μ l−1. Haematological parameters: filgrastim (300 mcg per day) was started on day 4 and continued until WBC>3. To start each cycle, it was necessary to have neutrophils>1 × 109 l−1 and platelets>60 × 109 l−1. Renal parameters: U and E and creatinine (Cr) were measured daily, and if Cr rose >20%, no cisplatin was given until Cr reduced below that level. If the serum creatinine rose clearance by 15%, an EDTA clearance was repeated. If the clearance was <40 ml min−1, carboplatin AUC 4 was substituted for cisplatin. In this case, vincristine was given on weeks 2, 4, 7 and 9. If the clearance subsequently improved, then cisplatin was reintroduced and vincristine was dropped. Dose reductions: a 20% dose reduction was made in the doses of cisplatin, actinomycin-D and etoposide, in the presence of platelets<20 × 109 l−1 or neutrophils<0.5 × 109 l−1 for>5 days. A 20% reduction in the dose of MTX was made in the presence of grade 3 or 4 mucositis. A 50% dose reduction in the dose of MTX was made, if the 24-h level was >80 μ l−1 or the 48 h level was >4 μ l−1.
An induction cycle of baby BOP (cisplatin 50 mg m−2, vincristine 2 mg and bleomycin 30 000 U over 12 h) was given to patients who had renal obstruction, respiratory failure due to disease, extensive inferior vena cava thrombosis thought to be at high risk of pulmonary embolism and patients with poor performance status (Eastern Co-operative Oncology Group PS 3). In these individuals, GAMEC was given 10–14 days later with omission of cycle 1 day 8 cisplatin. Drug doses were calculated on the basis of body surface area; the full dose was deemed to be 100%, if a 20% dose reduction was needed then the dose was said to be 80%. This percentage dose was multiplied by the dose delivery (actual inter-cycle length divided by intended inter-cycle length expressed as a percentage). The drugs were analysed separately and then combined, assuming the contribution of each drug to be equal.
At the end of therapy, patients underwent surgery to remove sites of disease greater than 1 cm in diameter (3 cm in seminoma). If any viable germ cell tumour elements were found, no further therapy was given, unless the patient relapsed. In patients with brain metastases, radiotherapy was not routinely administered. Patients who progressed subsequently were offered further chemotherapy with high-dose chemotherapy consolidation.
Radiological assessment was carried out before week 6, unless tumour markers showed a response. Treatment was not stopped if tumour markers failed to decline at anticipated half-life. Tumour markers including alpha foetoprotein (AFP) and human chorionic gonadotrophin (βHCG) were routinely followed. In some patients, the AFP rose during therapy, while the patient was responding due to AFP production by the liver and additional scanning was used to clarify this situation.
Pharmacokinetics
Twenty-four EDTA blood samples were collected over a 72 h time period from the start of treatment. Plasma was separated by centrifugation (1200 g for 10 min) and stored at −40°C until analysis. Methotrexate, 7-hydroxymethotrexate (7-OH MTX) and etoposide were detected by two separate HPLC methods with UV detection. Actinomycin-D was measured by a novel LC-MS-MS method with a sensitivity of <1 ng ml−1 (Veal et al, 2005). PK analysis was carried out in Kinetica (Innaphase Corp, Philadelphia, PA, USA) using non-compartmental methods. Area under the concentration–time curve (AUC) was determined by the trapezoidal method, using the linear rule for ascending concentrations and the log linear rule for descending concentrations. Area under the concentration–time curve was extrapolated to infinity by dividing the concentration at 24 h by the elimination rate constant (λz), to derive AUC0–∞. Total plasma clearance was derived from the dose divided by AUC(0−∞), or from steady-state concentration at 12 h (CSS) divided by the infusion rate (mg h−1). Elimination half-life (t1/2) was calculated as 0.693/λz.
Statistical methods
Statistical analyses were performed with the STATA 8.2 statistical software package. Overall survival (OS) and PFS were assessed using the Cox proportional hazard method for continuous variables. Survival curves were generated using the Kaplan–Meier method. Categorical variables were analysed using the log-rank test. To assess the correlation between the various categorical variables and being progression free (PF) to GAMEC, Fisher's exact test was used. The median test was used to examine continuous variables against PF status. Logistic regression was used to determine which factors were predictive of being PF. PK between group comparisons were made using the Mann–Whitney U-test. Progression-free and overall survivals were analysed on an intention to treat basis.
Results
The majority of patients had non-seminomatous GCT and the median age measured was 33 years (Table 1). At a median follow-up of 2.5 years, 20 (74%) untreated patients were PF following GAMEC and appropriate surgery (Table 2; Figure 2). There were two treatment-related deaths (TRDs), one additional patient was salvaged by further therapy (78% overall). Out of five patients with central nervous system metastases at presentation, three remain PF.

Overall survival.
Eighteen (51%) of the pretreated group were similarly PF and a further four were salvaged by additional therapy (63% overall). There were three TRDs. Nine pretreated patients had late relapses before GAMEC and five remain PF. Three patients had brain metastases on relapse, two had post-chemotherapy surgery confirming CR and that they were PF and one progressed and died. Eight patients had viable cancer at surgery, five of them had greater than 10% viable tumour and all five relapsed. The remaining three with less than 10% viable tumour were all PF.
High-dose chemotherapy was offered to 10 patients. In the untreated group, four received high-dose therapy, but it was unsuccessful in all of them. One received high-dose carboplatin AUC20 with etoposide 1600 mg m−2 and cyclophosphamide 6 g m−2 Another patient received high-dose carboplatin AUC20 with thiotepa 500 mg m−2 and cyclophosphamide 6 g m−2. A third patient received carboplatin AUC10 with irinotecan 200 mg m−2 and gemcitabine 360 mg m−2 over 72 h, supported by autologous stem cells and repeated and given sequentially three times. The fourth patient received high-dose carboplatin AUC20 with etoposide 1600 mg m−2 and melphalan 140 mg m−2. In the pretreated group, high-dose therapy consisted of high-dose carboplatin AUC20 with etoposide 1600 mg m−2 and cyclophosphamide 6 g m−2 in two patients and was successful in one. High-dose carboplatin AUC30 and etoposide 1600 mg m−2 was offered to one patient, it but resulted in a TRD. High-dose topotecan 30 mg m−2, carboplatin AUC21 and thiotepa 500 mg m−2 was offered to three patients as part of a clinical trial, and it was successful in two.
Toxicity
The most common toxicities were myelosuppression and mucositis (Table 3a). There were five TRDs (8%); four of these were due to sepsis and one was due to intra-abdominal haemorrhage from choriocarcinoma. A large number of cycles were complicated by febrile neutropenia, with about half of them requiring platelet transfusion (Table 3b).
Significant reversible renal dysfunction occurred in five of the untreated patients (grade 2 WHO criteria), of whom one was dialysed for 24 h following an episode of septic shock. His renal function before this had been normal and returned subsequently to normal. In the pretreated group, four were similarly affected, one of whom (who had significant renal problems with BEP) became dialysis dependent long term. A total of 6% of cycles required the omission of cisplatin, and in one-third of these cycles carboplatin was substituted. One patient required carboxypeptidase to inactivate MTX during the first cycle of treatment; his EDTA clearance before HD MTX had been normal and he did not receive any cisplatin on that cycle. Methotrexate was cautiously reintroduced on the third cycle without adverse effects after his renal impairment had subsided.
Two patients developed typhlitis, one required a defunctioning colosostomy that was subsequently reversed. Three patients developed thrombo-embolic disease (all central access line associated). Two patients required parenteral feeding. One patient developed transient occipital blindness, he had received two lines of prior cisplatin-based therapy. An MRI scan confirmed white-matter changes in the occipital lobe, thought to be due to cisplatin and not metastases. His vision subsequently recovered without further therapy.
Prognostic factors
We looked at the following prognostic factors to see if they correlated with PF status. They were age, presence of non-seminomatous histology, raised lactate dehydrogenase (LDH), Memorial Sloan-Kettering Cancer Center (MSKCC) criteria and Medical Research Council (MRC) UK criteria for relapsed GCT outcome. Only age (<median age vs >median age: 72 vs 29%, FET (Fisher's exact test)=0.018) and raised LDH before GAMEC (normal LDH vs raised LDH: 67 vs 29%, FET=0.041) were significant. In the pretreated group, neither the MRC criteria for adverse outcome (βHCG or αFP >100, failure to achieve a CR to initial chemotherapy, relapse within 2 years) (Fossa et al, 1999) nor those developed by MSKCC (Motzer et al, 2000), including extragonadal primary, failure to achieve at least marker-negative partial response (m−ve PR), failure of two lines of cisplatin-based therapy), were able to define a poor-prognosis group. The good-risk group defined by the MSKCC (gonadal primary, one line of cisplatin-based therapy, achievement of at least m−vePR) (7/14 PF) and relapse at least 6 months from the end of the last chemotherapy, also failed to define a group with a better prognosis (8/12 PF). Only age and raised LDH were significant on multivariate analysis (Figures 3, 4 and 5).

PFS in pretreated patients by absence or presence of raised LDH.

PFS in pretreated patients by age (above and below median).

PFS by Memorial Sloan-Kettering Risk Group.
Dose delivery and outcome
In the untreated group, no relationship between dose delivered or dose density was seen. In the pretreated group, when the two components of dose density were entered as separate variables into a regression model, both dose and inter-cycle delay were found to be independently associated with PFS (Figure 6). The ideal (100% dose) for MTX was adjusted for renal function, as shown in Figure 1. The number of patients maintaining the dose of MTX at ⩾80% over the first 2 cycles was also significant; no other drug alone showed a significant effect. However, for the four drugs together, the overall dose density delivered was highly significant (Table 4). Beyond the first two cycles, the effect of dose density was no longer statistically significant for PFS.

PFS in pretreated patients by dose density over cycles 1 and 2.
Pharmacokinetics and pharmacodynamics
Pharmacokinetic data was obtained on 43 patients for MTX and etoposide, and 31 patients for actinomycin-D. Summary data are presented in Table 5. There was no relationship between renal function (serum creatinine) and plasma clearance of MTX, actinomycin-D or etoposide (r2<0.1), although only seven patients had serum creatinine values above the normal range (>115 μmol l−1), and all were <139 μmol l−1.
The main aim of the pharmacological aspect of the study was to identify relationships between pharmacokinetic parameters and pharmacodynamic effects. The primary pharmacodynamic measures were PF status for efficacy, and neutrophils <0.5 × 109/l, for <4days or ⩾4 days for toxicity. AUC0−∞ was taken as the primary PK measure. methotrexate, actinomycin-D or etoposide AUC were not significantly predictive of efficacy or toxicity (P>0.1 throughout), nor was MTX CSS at 12 h. 7-Hydroxymethotrexate AUC0−∞ was not predictive of renal toxicity, as indicated by a >30% increase in serum creatinine (931±518 vs 761±468 μg ml−1h−1 in patients with and without renal toxicity, respectively, P>0.1).
As an overall measure of total cytotoxic exposure, the AUC values for each drug were normalised (AUC/mean AUC) and summed up. These values were also not predictive of efficacy (1.9±0.3 vs 2.0±0.7, P>0.1) or toxicity (1.9±0.7 vs 2.0±0.4, P>0.1) for MTX+etoposide or for MTX+etoposide +actinomycin-D (2.9±0.4 vs 3.0±1.2 and 2.8±1.1 vs 2.8±0.6, respectively, P>0.1). Grouping patients according to individual PK values being greater or less than the median values suggested an increased likelihood of treatment failure in patients with MTX AUC values <median (13/21 vs 7/22, P=0.048).
Discussion
We show that the novel regimen GAMEC, with appropriate surgery, produces a PFS of 74% in untreated patients and 51% in previously treated patients. These data suggest this treatment to be highly active in GCT. The permissive entry criteria allowing patients without formal histology to enter the study meant that very ill patients could be treated using this protocol. In addition, the use of an induction cycle of baby BOP allowed these patients to be stabilised before commencing intensive therapy, and the use of HD MTX here negated the requirement for cranial irradiation.
Many studies for intensive protocols in untreated GCT have employed large doses of bleomycin, making them unsuitable for salvage treatment. This regimen did not include bleomycin. Concerns that this might compromise outcome could be off-set by the fact that actinomycin-D produces a response rate of 38%, which is greater than that of bleomycin (Early and Albert, 1976).
The regimen was accompanied by significant toxicity, with five TRDs. These patients were older and one had only recently been exubated following a laparotomy, and was severely malnourished.
There was a high frequency of mucositis and febrile neutropenia. Many cycles required the use of platelets, particularly if the patients had been pretreated (Table 3b). It proved difficult to deliver the treatment in older patients (>35 years), and this was the probable reason for their inferior survival. Nevertheless, for those who relapsed, it was possible to give further therapy to all of the patients in the untreated group and to the vast majority (16/17) of the pretreated group. Adequate stem cell collections were also possible in 9/11 patients in whom it were attempted, suggesting that deferring high-dose chemotherapy for patients who relapsed after GAMEC was practical.
We have shown that it is possible to deliver HD MTX in combination with cisplatin (cisplatin given 38 h after HD MTX). The measurement of the serum creatinine 24 h post-MTX allowed identification of patients who had developed renal impairment, and future chemotherapy could be dosed accordingly. The results of the pharmacokinetic studies lead to the conclusion that 7-OH MTX is not the cause of MTX-induced renal dysfunction, as has been widely held to be the case. In these patients, cisplatin was withheld until the serum creatinine fell, with levels checked every 12 h. The role of MTX in relapsed disease remains controversial, while single-agent data have proved to be disappointing. However, regimens incorporating MTX have proved to be encouraging. Two other intensive regimens have used MTX in intermediate doses (BOMP/EPI (Germa-Lluch et al, 1999) and POMB/ACE (Husband and Green, 1992)). Both used alkylating agents, although the dose in POMB/ACE was low. The single-agent data in relapsed disease used 1 g m−2 (Atkinson et al, 1987), substantially lower than that used here.
The median delivery of cisplatin was 250 mg m−2 in the pretreated and 270 mg m−2 in the untreated patients, suggesting the targeted delivery of 300 mg m−2 to be realistic. The relationship between dose density and outcome could only be found in the pretreated group. It appeared that maintenance of dose density over the first two cycles was the most important factor, with dose intensity and absence of dose delay being equally important. The failure to see such a relationship in the untreated group probably reflects the low number of relapses in this group in the first place, and this is similar to the finding with POMB/ACE chemotherapy (Husband and Green, 1992). The failure of double-dose cisplatin in BEP to improve survival in untreated patients questions the wisdom of sole escalation of cisplatin as a major strategy to improve outcome (Nichols et al, 1991).
For relapsed patients, the data presented are comparable to that from other groups using high-dose therapy (Bhatia et al, 2000; Motzer et al, 2000). Other groups have reported encouraging results using alternative agents, for instance cisplatin and epirubicin (Bedano et al, 2005), and recently an interesting case report suggested that antiangiogenic agents may also have a role (Voigt et al, 2006).
We failed to detect a significant difference in the outcome of these patients, when the two most commonly used scoring systems for prognosis (MRC (Fossa et al, 1999) and MSKCC (McCaffrey et al, 1997)) were applied. It appears that the good group (using the MSKCC criteria), for whom it has been suggested that the cisplatin, ifosfamide and paclitaxel (TIP)-based approach is frequently curative, fare as well as those reported (67% with GAMEC vs 65%) (Kondagunta et al, 2005). Results from the poor-risk relapse group, for whom conventional therapy seems inadequate, appear as good as those reported for ifosfamide/paclitaxel induction followed by three cycles of high-dose carboplatin and etoposide treatment, supported by autologous blood stem cells (50% with GAMEC vs 41%) (Motzer et al, 2000).
We could only identify two prognostic factors that correlated with poor outcome, namely age and raised LDH before GAMEC. It might have been that these simply were the patients who received a lower intensity of therapy. In fact all of the six patients with raised LDH who received <80% dose density over the first two cycles, relapsed vs four of eight of those who received the higher dose intensity. A similar finding existed for age (2/10 vs 3/6). Although not statistically significant, there is a suggestion that adequate dose intensity may overcome these adverse prognostic factors.
Late relapses appear salvageable with GAMEC (5/9), a group thought to be chemorefractory. Recently, a similar finding has been described with TIP (7/14) (Ronnen et al, 2005). Derived PK values for MTX, actinomycin-D and etoposide clearance were inline with previously published values (Clark et al, 1994; Crews et al, 2004; Veal et al, 2005). These were not predictive of outcome. The use of multiple drugs was a confounding factor, especially as cisplatin pharmacokinetics were not assessed. The use of filgrastim and folinate will have reduced the impact of pharmacokinetic variability on myelosupression. Despite these caveats, there was a trend towards lower MTX AUC in patients who relapsed.
Overall, these results show that GAMEC is an effective therapy both for untreated patients and those who relapse. Further relapses can still receive treatment thereafter. The data suggest that the current prognostic scoring systems for relapsed patients fail to identify a poor-prognosis group for this regimen. These encouraging results have been obtained with established agents.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Anthoney DA, McKean MJ, Roberts JT, Hutcheon AW, Graham J, Jones W, Paul J, Kaye SB (2004) Bleomycin, vincristine, cisplatin/bleomycin, etoposide, cisplatin chemotherapy: an alternating, dose intense regimen producing promising results in untreated patients with intermediate or poor prognosis malignant germ-cell tumours. Br J Cancer 90: 601–606
Atkinson CH, Horwich A, Peckham MJ (1987) Methotrexate for relapse of metastatic non-seminomatous germ-cell tumours. Med Oncol Tumor Pharmacother 4: 33–37
Bedano P, Brames M, Williams S, Einhorn L (2005) A phase II study of cisplatin plus epirubicin salvage chemotherapy in refractory germ cell tumors. J Clin Oncol 23, ASCO Abstract, 4526
Bhatia S, Abonour R, Porcu P, Seshadri R, Nichols C, Cornetta K, Einhorn L (2000) High dose chemotherapy as initial salvage chemotherapy in patients with relapsed testicular cancer. J Clin Oncol 18: 3346–3351
Bower M, Newlands ES, Holden L, Rustin GJ, Begent RH (1997) Treatment of men with metastatic non-seminomatous germ cell tumours with cyclical POMB/ACE chemotherapy. Ann Oncol 8: 477–483
Christian JA, Huddart RA, Norman A, Mason M, Fossa S, Aass N, Nicholl EJ, Dearnaley DP, Horwich A (2003) Intensive induction chemotherapy with CBOP/BEP in patients with poor prognosis germ cell tumors. J Clin Oncol 21: 871–877
Clark PI, Slevin ML, Joel SP, Osborne RJ, Talbot DI, Johnson PW, Reznek R, Masud T, Gregory W, Wrigley PF (1994) A randomized trial of two etoposide schedules in small-cell lung cancer: the influence of pharmacokinetics on efficacy and toxicity. J Clin Oncol 12: 1427–1435
Crews KR, Liu T, Rodriguez-Galindo C, Tan M, Meyer WH, Panetta JC, Link MP, Daw NC (2004) High-dose methotrexate pharmacokinetics and outcome of children and young adults with osteosarcoma. Cancer 100: 1724–1733
Early KS, Albert DJ (1976) Single agent chemotherapy (actinomycin D) in the treatment of metastatic testicular carcinoma. South Med J 69: 1017–1021
Evans WE, Relling MV, Rodman JH, Crom WR, Boyett JM, Pui CH (1998) Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med 338: 499–505
Fossa S, Stenning S, Gerl A, Horwich A, Clark P, Wilkinson P, Jones W, Williams M, Oliver R, Newlands E, Mead G, Cullen M, Kaye S, Rustin G, Cook P (1999) Prognostic factors in patients progressing after cisplatin-based chemotherapy for malignant non-seminomatous germ cell tumours. Br J Cancer 80: 1392–1399
Germa-Lluch JR, Garcia del Muro X, Tabernero JM, Sanchez M, Aparicio J, Alba E, Barnadas A (1999) BOMP/EPI intensive alternating chemotherapy for IGCCC poor-prognosis germ-cell tumors: the Spanish Germ-Cell Cancer Group experience (GG)[see comment]. Ann Oncol 10: 289–293
Hinton S, Catalano PJ, Einhorn LH, Nichols CR, David Crawford E, Vogelzang N, Trump D, Loehrer Sr PJ (2003) Cisplatin, etoposide and either bleomycin or ifosfamide in the treatment of disseminated germ cell tumors: final analysis of an intergroup trial. Cancer 97: 1869–1875
Horwich A, Shipley J, Huddart R (2006) Testicular germ-cell cancer. Lancet 367: 754–765
Husband DJ, Green JA (1992) POMB/ACE chemotherapy in non-seminomatous germ cell tumours: outcome and importance of dose intensity. Eur J Cancer 28: 86–91
International Germ Cell Cancer Collaborative Group: (1997) a prognostic factor-based staging system for metastatic germ cell cancers. J Clin Oncol 15: 594–603
Kaye S, Mead G, Fossa SD, Cullen M, deWit R, Bodrogi I, Groeningen Cv, Sylvester R, Collette L, Stenning S, Prijk LD, Lallemand E, DeMulder P (1998) Intensive induction-sequential chemotherapy with BOP/VIP-B compared with treatment of metastatic nonseminomatous germ cell tumour. J Clin Oncol 16: 692–701
Kondagunta GV, Bacik J, Donadio A, Bajorin D, Marion S, Sheinfeld J, Bosl GJ, Motzer RJ (2005) Combination of paclitaxel, ifosfamide, and cisplatin is an effective second-line therapy for patients with relapsed testicular germ cell tumors. J Clin Oncol 23: 6549–6555
Levi JA, Thomson D, Harvey V, Gill G, Raghavan D, Tattersall M, Snyder R, Burns I, Sandeman T, Byrne M et al (1990) Effective salvage chemotherapy with etoposide, dactinomycin, and methotrexate in refractory germ cell cancer. Australasian Germ Cell Trial Group. J Clin Oncol 8: 27–32
McCaffrey J, Mazumdar M, Bajorin D, Bosl G, Vlamis V, Motzer R (1997) Ifosfamide and cisplatin containing chemotherapy as first line salvage threrapy in germ cell tumours response and survival. J Clin Oncol 15: 2559–2563
Mead GM, Cullen MH, Huddart R, Harper P, Rustin GJ, Cook PA, Stenning SP, Mason M, Party MRCTTW (2005) A phase II trial of TIP (paclitaxel, ifosfamide and cisplatin) given as second-line (post-BEP) salvage chemotherapy for patients with metastatic germ cell cancer: a medical research council trial. Br J Cancer 93: 178–184
Motzer RJ, Mazumdar M, Sheinfeld J, Bajorin DF, Macapinlac HA, Bains M, Reich L, Flombaum C, Mariani T, Tong WP, Bosl GJ (2000) Sequential dose-intensive paclitaxel, ifosfamide, carboplatin, and etoposide salvage therapy for germ cell tumor patients [see comment]. J Clin Oncol 18: 1173–1180
Nichols CR, Williams SD, Loehrer PJ, Greco FA, Crawford ED, Wettlaufer J, Miller ME, Bartolucci A, Schacter L, Einhorn LH (1991) Randomized study of cisplatin dose intensity in poor risk germ-cell tumours – a Southeastern cancer study group and Southwestern oncology group protocol. J Clin Oncol 9: 1163–1172
Rick O, Bokemeyer C, Beyer J, Hartmann JT, Schwella N, Kingreen D, Neureither S, Metzner B, Casper J, Wandt H, Hartmann F, Schmoll HJ, Derigs G, Gerl A, Berdel WE, Kanz L, Siegert W (2001) Salvage treatment with paclitaxel, ifosfamide, and cisplatin plus high-dose carboplatin, etoposide, and thiotepa followed by autologous stem-cell rescue in patients with relapsed or refractory germ cell cancer. J Clin Oncol 19: 81–88
Ronnen EA, Kondagunta GV, Bacik J, Marion S, Bajorin DF, Sheinfeld J, Bosl GJ, Motzer RJ (2005) Incidence of late-relapse germ cell tumor and outcome to salvage chemotherapy. J Clin Oncol 23: 6999–7004
Shamash J, Oliver R, Ong J, Raja M, Edmonds P, Gallagher C, Ostrowski M, LeVay J, Williams M (1999) 60% salvage rate for germ cell tumours using sequential m-BOP, surgery and ifosfamide based chemotherapy. Ann Oncol 10: 685–692
Toner GCSM, Boyer MJ, Jones M, Thomson DB, Harvey VJ, Olver IN, Dhillon H, McMullen A, Gebski VJ, Levi JA, Simes RJ (2001) Comparison of two standard chemotherapy regimens for good-prognosis germ-cell tumours: a randomised trial. Australian and New Zealand germ cell trial group. Lancet 357: 739–745
Veal GJ, Cole M, Errington J, Parry A, Hale J, Pearson AD, Howe K, Chisholm JC, Beane C, Brennan B, Waters F, Glaser A, Hemsworth S, McDowell H, Wright Y, Pritchard-Jones K, Pinkerton R, Jenner G, Nicholson J, Elsworth AM, Boddy AV, Kingdom Children's Cancer Study Group Pharmacology Working Group (2005) Pharmacokinetics of dactinomycin in a pediatric patient population: a United Kingdom Children's Cancer Study Group Study. Clin Cancer Res 11: 5893–5899
Voigt W, Kegel T, Maher G, Jordan K, Muller L, Schmoll HJ (2006) Bevacizumab plus high-dose ifosfamide, etoposide and carboplatin (HD-ICE) as third-line salvage chemotherapy induced an unexpected dramatic response in highly platinum refractory germ-cell cancer. Ann Oncol 17: 531–533
Acknowledgements
The funding for the pharmacokinetic studies and data management was provided through a grant from The Orchid Cancer Appeal. We would like to thank H Irwin, L Lundholm and E Coles for carrying out the drug quantitation assays, and also thank the members of the Anglian Germ Cell Collaborative Group (AGCCG) for referring patients for this study, and the nursing staff on Gordon Hamilton Fairley Ward, St Bartholomew's Hospital, London, UK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Shamash, J., Powles, T., Ansell, W. et al. GAMEC – a new intensive protocol for untreated poor prognosis and relapsed or refractory germ cell tumours. Br J Cancer 97, 308–314 (2007). https://doi.org/10.1038/sj.bjc.6603865
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603865
Keywords
This article is cited by
-
Accelerated BEP: a phase I trial of dose-dense BEP for intermediate and poor prognosis metastatic germ cell tumour
British Journal of Cancer (2011)
-
Malignant germ cell tumours in the elderly: a histopathological review of 50 cases in men aged 60 years or over
Modern Pathology (2008)
-
A novel treatment regimen for poor-prognosis or relapsed germ-cell tumors
Nature Clinical Practice Oncology (2007)
-
Conservative management of testicular germ-cell tumors
Nature Clinical Practice Urology (2007)
