
Abstract
Dendritic cells play a major role in the generation of immunity against tumour cells. They can be grown under various culture conditions, which influence the phenotypical and functional properties of dendritic cells and thereby the consecutive immune response mainly executed by T cells. Here we discuss various conditions, which are important during generation and administration of dendritic cells to elicit a tumouricidal T cell-based immune response.
Similar content being viewed by others
Main
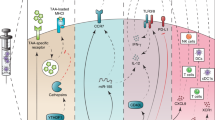
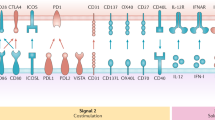
Cytotoxic T-cells (CTL) are the most efficient cells concerning defence against tumour cells (Lanzavecchia, 1993). One of the major questions in tumour immunology is, how an efficient CTL-response can be generated to elicit an antigen-specific and protective T-cell response in vivo. It has been shown that the immune response to tumour antigens and other antigens is altered in patients with cancer. These alterations concern many elements of the immune response and prevent effective proliferation of tumour-antigen specific T cells and their subsequent recognition of tumour cells. In recent studies antigen-presenting cells (APC) have been shown to play a crucial role in the induction of tumour-protective immune responses by generating tumour-specific T cells (Lanzavecchia, 1993). Antigen receptors (TCR) of tumour-specific T cells recognise tumour-associated peptides that are presented in the context of HLA class-I or class-II molecules by the APC. Successful recognition of tumour–antigen by the T-cell is not only dependent on TCR-peptide-HLA-interaction, but other co-stimulatory signals must be provided to prevent anergy (Schwartz, 1990). These are mainly CD80/CD86–CD28- or CD40–CD40L-interactions (Bennett et al, 1998). These interactions do not only underline the importance of T cells, but also the significant role of dendritic cells (DC), which are the most potent antigen-presenting cells among others like monocytes, macrophages and B cells.
Several in vitro and in vivo studies showed the ability of vaccination with DC to elicit tumour-specific T-cell immunity (Schuler and Steinman, 1997). This result implies that (1) DC might be just another altered element of the immune system or (2) DC are able to overcome tumour-protective alterations in cancer patients by inducing effective CTL response or (3) both. In this context a phenotypic and functional dichotomy of DC in DC1 and DC2 appears to be of importance. DC1 and DC2 cells were found to produce different cytokines and thereby induce TH1 and TH2 differentiation, respectively. The lymphoid-related DC (DC2) are CD11c− and have been shown to induce a tolerating response vs tumour cells by activating mainly TH2 cells, whereas myeloid-derived DC (DC1) are immunostimulatory via TH1 cells. Development of TH2-promoting DC2 cells is inhibited by cytokines (IL-4) produced by TH2 cells. In contrast, development of TH1-promoting DC1 cells is enhanced by the TH2 cytokine IL-4 (Banchereau and Steinman, 1998; Rissoan et al, 1999).
Vaccination with peptide- or lysate-pulsed DC showed the clinical efficiency in the induction of a curative tumour-specific therapy in metastatic melanoma and other malignancies (Table 1; Thurner et al, 1999a). In a study performed with peptide- and lysate-pulsed DC five out of 16 patients showed at least a partial remission, two of them a complete remission (Nestle et al, 1998). Monocytes drawn from peripheral blood were grown in the presence of a cytokine cocktail (GM–CSF, IL-4, IL-1β, IL-6, TNF-α, PgE2) to DC, which display mainly the DC1-phenotype. Subsequently, DC were pulsed with peptides or autologous tumour-lysate and injected back into the patient with different clinical outcome.
Several factors seem to influence a successful vaccination by peptide- or lysate-pulsed DC: (1) Generation of DC; (2) Selection of tumour-antigen pulsing of DC; (3) Transfection into DC; and (4) Route of application of DC.
Generation of DC
Physiologically, human DC are mainly localised in tissue and represent only a small portion of less than 0.5% of peripheral blood leukocytes. For therapeutical purposes large numbers of DC are needed.
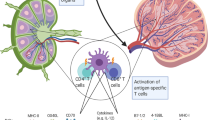
DC can either be generated from proliferating CD34+ bone marrow precursor cells (Caux et al, 1996) – which differentiate under a variety of different cytokines including SCF, Flt3, GM–CSF, TGF-β and TNF-α – or from non-proliferating peripheral CD14+ cells (monocytes) (Sallusto and Lanzavecchia, 1994). Usually, CD34+ precursors mobilised by G-CSF are isolated by leukapheresis to obtain high numbers of peripheral cells for therapeutical purposes. These cells seem to be more efficient in the activation of tumour-specific CTLs than CD14+ derived DC (Mortarini et al, 1997). CD34+ cells expand 10–30-fold. Yields of 5 × 106 cells per leukapheresis are typically obtained. In contrast, monocytes are abundantly present in peripheral blood and can be easily obtained by peripheral blood drawings or leukapheresis. Protocols for the generation of large amounts of monocyte-derived DC are known since 1994 (Romani et al, 1994; Sallusto and Lanzavecchia, 1994) and have been used for both experimental and therapeutical purposes. Here, leukocytes are prepared from peripheral blood using Ficoll–Hypaque density centrifugation. Monocytes are isolated by an adherence step and subsequently cultured in the presence of GM–CSF, IL-4 and 10% FCS or alternatively – under serum free conditions (Jonuleit et al, 1997; Thurner et al, 1999b) – with 1% autologous plasma for 7 days (Figure 1). After 1 week the yield of DC generated varies from about 25 to 50% of the starting population. Yields of 0.5–2.0 × 106 cells per 10 ml blood are typically obtained. Adherent cells show cytoplasmic processes typical for DC. After co-culturing with immunologic effector cells DC form typical cluster.

Schematic diagram of dendritic cell generation.
DC display several antigens on their cell surface, all of which are characteristic, but not specific. The most typical markers at present are HLA-class-I-, -class-II-molecules and co-stimulatory markers (CD80, CD86). Immature DC – obtained after 7 days of culture with GM–CSF and IL-4 – can be grown to mature DC by co-culturing with TNF-α, IL-6, IL-1β, and PGE2 or, alternatively, with a so-called monocyte conditioned medium (MCM) for another 3 days (maturation phase) (Thurner et al, 1999b). In contrast to immature DC, mature DC are much more potent in inducing TH1 and CTL responses in vitro and are resistant to immunosuppressive effects of tumour-derived IL-10 (Steinbrink et al, 1999). Therefore, mature DC have been used in recent vaccination protocols (Thurner et al, 1999a). Table 2 shows major phenotypical and functional differences between immature and mature DC.
Selection of tumour-antigen pulsing of DC
Small antigenic peptides (consisting of 8–10 amino acids) are loaded directly from outside the cell on HLA-class-I-molecules, whereas tumour-lysates – as a protein or oligopeptide (>10 amino-acids) – are internalised by endocytosis into the antigen presenting cell, processed and then presented with the HLA class-II-molecule. Cross-presentation of tumour-lysate is possible, i.e. the presentation of proteins or oligopeptides with HLA class-I-molecules (Bennett et al, 1997). This implies that pulsing with tumour-lysate or oligopeptides is also able to activate CD8+ T-cells immediately by the HLA-class-I-pathway and not only CD4+ T-cells by the ‘conventional’ HLA-class-II-pathway. CD40/CD40L signalling via DC/CD4+ T-cell-interaction is able to pre-activate DC temporarily. Subsequently these CD4+ T-cell-pre-activated DC can generate cytotoxic responses in CD8+ T cells (Bennett et al, 1998).
The selection of the peptide used for vaccination, is influenced by several factors: type of tumour, HLA class-I or -II of the patient, successful induction of CTL-response in vitro or in vivo, etc. Today, more than 50 melanoma-associated epitopes are known, which can be recognised by T cells. These epitopes are presented via different HLA class-I- and HLA class-II-molecules (e.g. HLA A1, HLA A2, HLA DR4, …). An optimal selection of these epitopes allows the treatment of almost 100% of patients with peptide-pulsed DC for the case of malignant melanoma.
Vaccination of tumour–patients with a single peptide can result in peptide-specific cytotoxicity. In these cases tumour escape mechanisms may be a problem, for example by the loss of tumour-associated epitopes or of essential antigen presenting molecules. This problem can be circumvented by the usage of polyvalent vaccines in a single patient, i.e. the application of tumour lysates or the application of several peptides. This phenomenon can also be observed in a subgroup of a trial conducted by Nestle et al (1998). Patients treated with tumour–lysate showed highest response rates (50%, two out of four) compared to patients treated with peptides alone (25%, three out of 12) (Table 1).
Tumour-associated peptides do bind with a defined affinity both to the HLA molecules (i.e. HLA class-I and HLA class-II) and the TCR. Whether this peptide is useful for therapeutical purposes or not mainly depends on the degree of its affinity. Low affinity to the HLA molecules is synonymous with low potency in CTL induction, whereas high affinity means high potency in CTL induction (Sette et al, 1994). Therefore, vaccination was performed with a so-called heteroclitic peptide. Original melanoma-associated peptides show a substitution of one amino acid with another at the same position. Heteroclitic peptides, that are changed at the HLA-binding motif to achieve a higher affinity between the HLA and the peptide, are potent immunogens. They are able to elicit cross-reactivity with the original peptide, because the TCR-binding motif remains unchanged. As a consequence a tumour-protective immune response against the original peptide can occur after vaccination with the heteroclitic peptide (Rosenberg et al, 1998).
Transfection into dendritic cells
Enhancing the immunogenicity of tumour cells is an interesting approach to cancer gene therapy (Schmidt-Wolf et al, 1994). Cytokine genes have been used in most instances to enhance tumour immunogenicity (Schmidt-Wolf and Schmidt-Wolf, 1996). DC are attractive targets of gene transfer since DC are easily accessable and since these cells seem to be sensitive to immunologic strategies. For further enhancement of the antigeneic presentation by DC various genes like the genes for interleukin-7 (Westermann et al, 1998), GM–CSF, interleukin-12, interferon-gamma and interferon-alpha (Tüting et al, 1998) have been transfected into DC. Up to 10% transfection efficiencies using electroporation for gene transfer into CD83+ mononuclear cell derived DC were reported. Other non-viral techniques produce robust DC transfection with 17% of monocyte-derived DC using cationic peptide or report the ability of using lipofection in principle.
Higher efficiencies can be achieved using viral vectors. Adenoviral vectors seem to be the most efficient transfection method (Mulders et al, 1998). Fifty to 85% transfected CD83+ DC generated from PBMC were reported. Thirty to 40% of precursor DC derived from human umbilical cord blood can be transduced using adenoviral vectors without cytopathic effect. With the aid of liposome-mediated infection, gene transfer into CD83+ DC resulted in more than 90% of the cells transduced. Using a protocol with UV-irradiated adenoviruses similar results can be obtained without addition of liposomes (Mulders et al, 1998; Märten et al, 2001). Adenoviral vectors can also be used for transduction of CD34+ cell derived DC (Bregni et al, 1998). For retroviral vectors a transduction efficiency of 10–30% has been reported; other groups described a resistance of DC to transduction by retroviral vectors. Recently, there were reports of using other viral vectors like fowlpox virus, lentivirus, avipoxvirus or vaccinia virus (Di Nicola et al, 1998).
Route of application of DC
Cell-based immunotherapy strategies using peptide- or lysate-pulsed DC require interaction between DC and T cells. Physiologically, bone marrow-derived DC or their progenitors migrate to tissues of inflammation, internalise antigens and subsequently reach the paracortex of the lymph nodes (Steinman, 1991) and the periarteriolar lymphoid sheath of the spleen (PALS). Here, DC prime naive T cells. The optimal route of administration of in vitro-cultured DC for migration to T-cell-rich sites is unknown, particularly since migratory capacity of cultured tumour-antigen-pulsed DC may be altered. Possible routes of administration are intradermal, subcutaneous, intranodal, intravenous and intraperitoneal injection of DC. Except intraperitoneal injection, which was performed in animal studies only, all of these applications have been employed in human cell-based vaccination protocols (Table 1). To examine migration patterns of DC, they were radioactively labelled with indium-111. Subsequently, tumour antigen-pulsed DC were administered by an intravenous, subcutaneous, or intradermal injection in patients with metastatic malignancies (Morse et al, 1999): Three patients received intravenous injection, four patients received intradermal or subcutaneous injections simultaneously on both sides of their body. Intravenous injection revealed highest activity in the lungs after 1 min, which decreased continuously and redistributed after 24 h to highest spleen and liver activities. No activity was found in lymph nodes and tumour. Subcutaneous injection showed no activity in the lymph nodes. It could not be detected where the injected DC remained. Intradermal injection revealed highest activity in the draining lymph nodes after 24 h. Only 0.1–0.4% of relative activity was found here, i.e. only 4000 of 106 injected cells reached the lymph node in contrast to 106 of 106 cells after successful intranodal injection. In conclusion, greatest activity in lymph nodes was only found after intradermal injection. Subcutaneous injection seemed to be ineffective and intravenous injection showed accumulation in the spleen as a T-cell-rich area. Technetium-labelled immature monocyte-derived DC have also been shown to migrate rapidly to the draining lymph nodes after intradermal injection (Thomas et al, 1999). Nonetheless, injecting DC directly into the lymph node seemed to deliver highest numbers of DC in T-cell-rich area, although this mode of application may destroy the normal architecture of the lymph node.
Studies where antigens were continuously injected into a lymph node, for example by a pump, have yet to be performed in humans. It has been shown that route and kinetic of peptide administration determine its immunogenicity. This may also be the case for DC administration.
Adverse effects
Peptide- or tumour-lysate pulsed DC are able to induce CTL-response in patients with malignant melanoma. Peptide or tumour-lysate antigens, used for DC vaccination, are normally not restricted to tumour tissue, but can be found at least partially on healthy tissue. Therefore a risk for the development of autoimmune diseases exists, that has been shown in animal models (Ludewig et al, 2000). Pilot clinical studies in humans could not find clinical signs of auto-immunity except vitiligo and the occurrence of auto-antibodies (anti-TSH-receptor-Ab, ANA). Unexpectedly, vaccination with tumour-lysate pulsed DC did not show a higher incidence of auto-immunity than vaccination with peptide-pulsed DC. The occurrence of IgG, IgM and IgE antibodies to bovine serum albumin (BSA) causing anaphylaxis after vaccination with human peptide-pulsed DC was reported (Mackensen et al, 2000). Therefore, for therapeutical ex vivo applications the use of serum-free generated DC was recommended.
More frequently systemic flu-like symptoms occurred: fever or painful swelling of the injected lymph node (after intranodal injection). After intradermal injection of the pulsed DC swelling, itching and erythema at the injection site could be detect. These reactions regressed within 48–72 h.
Induction of tolerance against tumour cells may be a problem, although recent studies pointed out the possible therapeutical value of DC vaccination.
Future developments
In conclusion, DC are able to increase the tumouricidal activity of immunologic effector T cells against tumour cells. This ability depends on several factors as has been discussed. Nonetheless, factors that influence effectively DC-activation of T cells against tumour cells have to be optimised. The generation of DC-subtypes that are more effective than those known today seems to be promising in inducing tumouricidal, specific immune responses not only in patients with malignant melanoma, but other malignant diseases. DC-based cell therapy will not only be conducted in patients with distant metastases, but also in patients with minimal residual disease or in adjuvant settings for high risk situations, where tumour-load is low and therefore tumours can be recognised specifically and eliminated effectively by DC-activated immunologic effector cells.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392: 245–252
Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR (1998) Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393: 478–480
Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR (1997) Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med 186: 65–70
Bregni M, Shammah S, Malaffo F, Di Nicola M, Milanesi M, Magni M, Matteucci P, Ravagnani F, Jordan CT, Siena S, Gianni AM (1998) Adenovirus vectors for gene transduction into mobilized blood CD34+ cells. Gene Ther 5: 465–472
Caux C, Vanbervliet B, Massacrier C, Dezutter-Dambuyant C, de Saint-Vis B, Jacquet C, Yoneda K, Imamura S, Schmitt D, Banchereau J (1996) CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF+TNF alpha. J Exp Med 184: 695–706
Di Nicola M, Siena S, Bregni M, Longoni P, Magni M, Milanesi M, Matteucci P, Mortarini R, Anichini A, Parmiani G, Drexler I, Erfle V, Sutter G, Gianni AM (1998) Gene transfer into human dendritic antigen-presenting cells by vaccinia virus and adenovirus vectors. Cancer Gene Ther 5: 350–356
Fong L, Brockstedt D, Benike C, Breen JK, Strang G, Ruegg CL, Engleman EG (2001) Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol 167: 7150–7156
Geiger JD, Hutchinson RJ, Hohenkirk LF, McKenna EA, Yanik GA, Levine JE, Chang AE, Braun TM, Mule JJ (2001) Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res 61: 8513–8519
Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R (1996) Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med 2: 52–58
Hu X, Chakraborty NG, Sporn JR, Kurtzman SH, Ergin MT, Mukherji B (1996) Enhancement of cytolytic T lymphocyte precursor frequency in melanoma patients following immunization with the MAGE-1 peptide loaded antigen presenting cell-based vaccine. Cancer Res 56: 2479–2483
Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH (1997) Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol 27: 3135–3142
Lanzavecchia A (1993) Identifying strategies for immune intervention. Science 260: 937–944
Ludewig B, Ochsenbein AF, Odermatt B, Paulin D, Hengartner H, Zinkernagel RM (2000) Immunotherapy with dendritic cells directed against tumor antigens shared with normal host cells results in severe autoimmune disease. J Exp Med 191: 795–804
Märten A, Ziske C, Schöttger B, Weineck S, Buttgereit P, Schakowski F, von Rücker A, Scheffold C, Sauerbruch T, Schmidt-Wolf I (2001) Transduction of dendritic cells (DC) with CIITA gene:Increase of immunostimulatory activity of DC. Cancer Gene Ther 8: 211–218
Mackensen A, Drager R, Schlesier M, Mertelsmann R, Lindemann A (2000) Presence of IgE antibodies to bovine serum albumin in a patient developing anaphylaxis after vaccination with human peptide-pulsed dendritic cells. Cancer Immunol Immunother 49: 152–156
Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK (1999) Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res 59: 56–58
Mortarini R, Anichini A, Di Nicola M, Siena S, Bregni M, Belli F, Molla A, Gianni AM, Parmiani G (1997) Autologous dendritic cells derived from CD34+ progenitors and from monocytes are not functionally equivalent antigen-presenting cells in the induction of melan-A/Mart-1(27-35)-specific CTLs from peripheral blood lymphocytes of melanoma patients with low frequency of CTL precursors. Cancer Res 57: 5534–5541
Mulders P, Pang S, Dannull J, Kaboo R, Hinkel A, Michel K, Tso CL, Roth M, Belldegrun A (1998) Highly efficient and consistent gene transfer into dendritic cells utilizing a combination of ultraviolet-irradiated adenovirus and poly(L-lysine) conjugates. Cancer Res 58: 956–961
Murphy G, Tjoa B, Ragde H, Kenny G, Boynton A (1996) Phase I clinical trial: T-cell therapy for prostate cancer using autologous dendritic cells pulsed with HLA-A0201-specific peptides from prostate-specific membrane antigen. Prostate 29: 371–380
Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D (1998) Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med 4: 328–332
Rissoan MC, Soumelis V, Kadowaki N, Grouard G, Briere F, de Waal M, Liu YJ (1999) Reciprocal control of T helper cell and dendritic cell differentiation. Science 283: 1183–1186
Romani N, Gruner S, Brang D, Kampgen E, Lenz A, Trockenbacher B, Konwalinka G, Fritsch PO, Steinman RM, Schuler G (1994) Proliferating dendritic cell progenitors in human blood. J Exp Med 180: 83–93
Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Dudley ME, Schwarz SL, Spiess PJ, Wunderlich JR, Parkhurst MR, Kawakami Y, Seipp CA, Einhorn JH, White DE (1998) Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 4: 321–327
Sallusto F, Lanzavecchia A (1994) Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med 179: 1109–1118
Schmidt-Wolf GD, Schmidt-Wolf IG (1996) Cancer and gene therapy. Ann Hematol 73: 207–218
Schmidt-Wolf IG, Huhn D, Neubauer A, Wittig B (1994) Interleukin-7 gene transfer in patients with metastatic colon carcinoma, renal cell carcinoma, melanoma, or with lymphoma. Hum Gene Ther 5: 1161–1168
Schott M, Seissler J, Lettmann M, Fouxon V, Scherbaum WA, Feldkamp J (2001) Immunotherapy for medullary thyroid carcinoma by dendritic cell vaccination. J Clin Endocrinol Metab 86: 4965–4969
Schuler G, Steinman RM (1997) Dendritic cells as adjuvants for immune-mediated resistance to tumors. J Exp Med 186: 1183–1187
Schwartz RH (1990) A cell culture model for T lymphocyte clonal anergy. Science 248: 1349–1356
Sette A, Vitiello A, Reherman B, Fowler P, Nayersina R, Kast WM, Melief CJ, Oseroff C, Yuan L, Ruppert J (1994) The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J Immunol 153: 5586–5592
Steinbrink K, Jonuleit H, Muller G, Schuler G, Knop J, Enk AH (1999) Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood 93: 1634–1642
Steinman RM (1991) The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9: 271–296
Thomas R, Chambers M, Boytar R, Barker K, Cavanagh LL, MacFadyen S, Smithers M, Jenkins M, Andersen J (1999) Immature human monocyte-derived dendritic cells migrate rapidly to draining lymph nodes after intradermal injection for melanoma immunotherapy. Melanoma Res 9: 474–481
Thurner B, Haendle I, Roder C, Dieckmann D, Keikavoussi P, Jonuleit H, Bender A, Maczek C, Schreiner D, von den Driesch P, Brocker EB, Steinman RM, Enk A, Kampgen E, Schuler G (1999a) Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med 190: 1669–1678
Thurner B, Roder C, Dieckmann D, Heuer M, Kruse M, Glaser A, Keikavoussi P, Kampgen E, Bender A, Schuler G (1999b) Generation of large numbers of fully mature and stable dendritic cells from leukapheresis products for clinical application. J Immunol Methods 223: 1–15
Timmermann JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, Taidi B, Rajapaska R, Caspar CB, Okada CY, van Beckhoven A, Liles TM, Engleman EG, Levy R (2002) Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune response in 35 patients. Blood 99: 1517–1526
Tüting T, Wilson CC, Martin DM, Kasamon YL, Rowles J, Ma DI, Slingluff CL, Wagner SN, van der Bruggen P, Baar J, Lotze MT, Storkus WJ (1998) Autologous human monocyte-derived dendritic cells genetically modified to express melanoma antigens elicit primary cytotoxic T cell responses in vitro: enhancement by cotransfection of genes encoding the Th1-biasing cytokines IL-12 and IFN-alpha. J Immunol 160: 1139–1147
Westermann J, Aicher A, Qin Z, Cayeux Z, Daemen K, Blankenstein T, Dorken B, Pezzutto A (1998) Retroviral interleukin-7 gene transfer into human dendritic cells enhances T cell activation. Gene Ther 5: 264–271
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Reinhard, G., Märten, A., Kiske, S. et al. Generation of dendritic cell-based vaccines for cancer therapy. Br J Cancer 86, 1529–1533 (2002). https://doi.org/10.1038/sj.bjc.6600316
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6600316
Keywords
This article is cited by
-
Enhanced immunity against hepatoma induced by dendritic cells pulsed with Hsp70-H22 peptide complexes and CD40L
Journal of Cancer Research and Clinical Oncology (2012)
-
Genomics and proteomics of immune modulatory effects of a butanol fraction of echinacea purpurea in human dendritic cells
BMC Genomics (2008)
-
Engineering of highly immunogenic long-lived DC vaccines by antiapoptotic protein gene transfer to enhance cancer vaccine potency
Gene Therapy (2008)
-
Human cytomegalovirus protein pp65: an efficient protein carrier system into human dendritic cells
Gene Therapy (2008)
-
A peptide-loaded dendritic cell based cytotoxic T-lymphocyte (CTL) vaccination strategy using peptides that span SIV Tat, Rev, and Env overlapping reading frames
Retrovirology (2006)
