
Abstract
Linkage disequilibrium (LD) structure is still unpredictable because the interplay of regional recombination rate and demographic history is poorly understood. We have compared the distribution of LD across two genomic regions differing in crossing-over activity – Xq13 (0.166 cM/Mb) and Xp22 (1.3 cM/Mb) – in 15 Eurasian populations. Demographic events predicted to increase the LD level – genetic drift, bottleneck and admixture – had a very strong impact on extent and patterns of regional LD across Xq13 compared to Xp22. The haplotype distribution of the DXS1225–DXS8082 microsatellites from Xq13 exhibiting strong association in all populations was remarkably influenced by population history. European populations shared one common haplotype with a frequency of 25–40%. The Volga-Ural populations studied, living at the geographic borderline of Europe, showed elevated LD as well as harboring a significant fraction of haplotypes originating from East Asia, thus reflecting their past migrations and admixture. In the young Kuusamo isolate from Finland, a bottleneck has led to allelic associations between loci and shifted the haplotype distribution, but has much less affected single microsatellite allele frequencies compared to the main Finnish population. The data show that the footprint of a demographic event is longer preserved in haplotype distribution within a region of low crossing-over rate, than in the information content of a single marker, or between actively recombining markers. As the knowledge of LD patterns is often chosen to assist association mapping of common disease, our conclusions emphasize the importance of understanding the history, structure and variation of a study population.
Similar content being viewed by others


Introduction
More than 10 years ago linkage disequilibrium (LD), or allelic association mapping, was pioneered as a tool for fine-scale localization of genes responsible for rare monogenetic diseases,1, 2 but has now come center-stage as the method of choice for finding the genes behind common diseases.3, 4 Both simulated as well as empirical data have shown that population history, in terms of bottlenecks, genetic drift in small populations, and admixture, has an impact on population background LD level.5 The data gathered from across different genomic regions also suggest the unique locus history of every genomic segment is influenced by local mutation, recombination rates and selection shaping the regional LD patterns.5, 6 Against the background of the recently initiated International HapMap project to create a genome-wide map of LD patterns in the human genome, there is still much debate as to whether this map would be applicable to association mapping in a population of interest or to the provision of detailed fine-scale structure for regions of interest.7, 8 More empirical data on worldwide populations are needed to understand how population history in combination with regional crossover activity acts on local LD patterns.
The X chromosome is a useful marker for population genetic studies owing to its intrinsic properties: accessible haplotypes in males, lower recombination rate, lower mutation rate and faster genetic drift due to smaller effective population size.9 As a result, we can expect LD to be greater on the X chromosome, and population structure more pronounced, compared to autosomes. In order to study the interplay between population history and recombination rate shaping the local LD patterns, we have chosen two X-chromosomal regions with contrasting crossing-over activity. Xq13 can be defined as an LD desert (0.166 cM/Mb), whereas Xp22 represents a recombination rate close to the average in human genome (1.3 cM/Mb). We have compared allelic associations for an extensive data set of 14 Eurasian populations and a further isolated subpopulation (including nine novel and six previously published populations). The populations have been chosen to represent different demographic scenarios predicted to generate LD: (i) the Saami and the Evenki: small constant populations, whose genepool has been influenced by genetic drift;10, 11 (ii) Kuusamo: a young, 300-years-old regional subpopulation of Finland, which has experienced multiple bottlenecks and is geographical isolated;12, 13 (iii) Volga-Ural populations of Mari, Udmurt, Chuvash and Komi: complex population history and ethnic structure due to geographic location on the borders of Europe and Asia.14 All the populations of the Volga-Ural region are distributed in smaller subgroups, often speaking distinct dialects, and having a wide geographic range even today. Based on mtDNA analysis the major part of the genepool of these populations is European.14, 15 The history of these populations, however, is rich in active contact with neighbors of different genetic background (East Asians, Turkic-speaking Bolgars, Tatars as well as Sub-Arctic groups).14, 16 Specifically, the Chuvash population is known from history to have arisen from the descendents of Turkic-speaking Volga Bolgars and local Fenno-Ugric tribes (apparently the Mari) after the fall of the Bolgar Kingdom due Mongol-Tatar invasion in around 1230. The Komi tribe is known to have moved to its current territories rich in taiga and tundra only after 900 AD. The founder population mixed with local Nenents tribes, as well as adopting their economy based on reindeer-breeding, hunting and fishing. The Mari have two subpopulations (eastern or meadow, and western or mountain Mari), speaking distinct dialects, and even with distinct written languages. For comparison we studied LD patterns for outbred and expanded populations of different size from eastern (Russians, Mordvin), northern (Finns, Estonians and Swedes), western (Dutch, Germans) and southern (Italians) Europe.
Materials and methods
Population samples
The sample sets of North-European DNA representing Finnish (n=80), Swedish (n=41), Estonian (n=45), the Saami, and the Evenki (n=71) populations are reported elsewhere.10, 11 The collection of the Volga-Ural population blood samples – Mari (n=44), Komi (n=46), Udmurt (n=49) and Chuvash (n=40) as well as Mordvin (n=49) – was conducted with help of local Blood Centers of Volga-Ural region and is described in detail by Bermisheva et al.15 The Russian DNA samples (n=66), collected in the framework of the International program INTAS Grant No. 93-0035, were kindly provided by Professor Voevoda (Institute of Internal Medicine, Novosibirsk, Russia). The German blood samples (n=41) represent the German population from the county of Munich (South-Bavaria) and were provided by Dr Weichhold (Institut für Rechtsmedicine, München). The Dutch blood samples (n=70) originating from the rural county of Doetinchem (Mid-East of Holland) were shared by Dr de Knijff. The Italian DNA samples (n=92) were kindly provided by Dr Cristian Capelli (The Forensic Genetics Laboratory, Istituto di Medicina Legale, Università Cattolica di Roma) and originate from the populations of Rome and Genova. The Kuusamo represents a recent isolate of north-eastern Finland, founded at the end of the 17th century by a nucleus of 39 families and now comprising 18 000 inhabitants that remained isolated until World War II.12 In this study the Kuusamo sample data were used as a model for recent bottleneck and rapid expansion. The East Asian Xq13 data set of Japanese and Mongolian populations described by Katoh et al17 was used as a reference source for east-Asian specific haplotypes. The Khalkh (1.8 million) represent the largest Mongolian population, whereas Khoton, Zahkchin and Uriankhai are young, isolated subpopulations (<25 000 people).
Laboratory procedures
All samples were genotyped for eight dinucleotide microsatellites on Xq13 and six markers on Xp22 selected from the Genethon genetic map as previously described.10, 11 All the genotypes were determined manually. The exact location (bp) on Human Genome Map of the microsatellites used is reported by Kaessmann et al.11 The order and distances in Mb/cM (deCode map) between the markers are the following:
-
1)
Xq13: DXS983 – 4.038/2.52 – DXS8037 – 0.731/<0.2 – DXS8092 – 7.183/<0.2 – DXS1225 – 0.162/<0.19 – DXS8082 – 1.053/<0.19 – DXS986 – 3.359/<1.83 – DXS1066 – 4.442/<1.83 – DXS995;
-
2)
Xp22: DXS987 – 1.627/<3.72 – DXS1053 – 3.723/<3.72 – DXS7163 – 0.814/<3.72 – DXS999 – 1.142/2.9 – DXS1229 – 1.901/5.39 – DXS989.
Data analysis
Arlequin version 2.0 software18 was used to estimate allele and haplotype frequencies, as well as calculate locus diversity (d) for each marker across both studied regions:  where pi is the estimated frequency of the ith allele at the locus. For each microsatellite locus in studied samples, the population mutation parameter θ=4Nμ (N is the effective population size; and μ the mutation rate) was estimated by maximum likelihood19 using MISAT software (http://www.bscb.cornell.edu/Homepages/Rasmus_Nielsen/files.html).
where pi is the estimated frequency of the ith allele at the locus. For each microsatellite locus in studied samples, the population mutation parameter θ=4Nμ (N is the effective population size; and μ the mutation rate) was estimated by maximum likelihood19 using MISAT software (http://www.bscb.cornell.edu/Homepages/Rasmus_Nielsen/files.html).
Using GOLD software,20 we calculated multiallelic extension of the normalized association measure D′ as

where p and q are observed allele frequencies at the two loci. As D′ is sensitive to rare allele frequencies, alleles with frequencies <10% were pooled. First, to address the level of background LD between unlinked markers, ‘baseline’ distribution of D′ values in each population sample was calculated for 48 possible pairs of unlinked microsatellites from Xq13 and Xp22. Second, we asked the question whether LD across studied regions is significantly different from the background LD between unlinked markers for each studied sample set. Mann–Whitney U-test was used to compare the distribution of D′ values between unlinked markers with the distribution of D′ values for marker-pairs across Xq13 or Xp22. Marker-pairs were grouped for the analysis according to distance separating them: (i) 0.1–2 Mb; (ii) 3–5 Mb; (iii) 5–10 Mb or (iv) 10–20 Mb.
Third, patterns of D′ values were used to compare LD levels across Xq13 among populations. However, as D′ estimates are strongly dependent on sample size,21, 22 we aimed to calculate D′ values for Xq13 marker-pairs for equally sized sample sets (n=40) from each population. Equal sample sizes were obtained by sampling of 40 random individuals from the original data sets of >40 individuals. Small sample size for Kuusamo (n=39) and Chuvash (n=40) did not allow resampling procedure. Sampling was repeated 100 times and D′ estimates were calculated as the mean of all replicates. Resampling in the present context was not meant as a traditional bootstrapping test to assess sample composition, but rather as an approach to achieve equal number of representative individuals from each studied population.
Significance of the allelic association between all possible locus pairs was also estimated by the tail probability (P-value) of Fisher's exact test, computed by the Genepop 3.0 software.23 For each pair of loci, r × c contingency table of gametes was formed and 1000 tables with the same marginal totals were generated based on a Markov chain algorithm. The procedure was repeated 500 times. The P-value is the mean fraction of such tables, which were equally or less likely than the observed table.
In order to compare the extent of overall LD across the studied regions, a multilocus LD statistic rd was computed for 10 Mb regions of Xp22 (all markers) and Xq13 (from DXS8037 to DXS986) using Multilocus 1.2 software (Paul-Michael Agapow and Austin Burt, http://www.bio.ic.ac.uk/evolve/software/multilocus). In essence, one is asking whether two individuals being the same at one locus makes them more likely to be the same at another. Rd is an extended statistic from traditional multilocus LD index of association, IA,24 correcting for the number of loci used in the analysis and thus making the comparison between different genomic regions possible. IA is based on comparing the variance of calculated pairwise differences between the haplotypes in the sample to the expected variance under the assumption of linkage equilibrium between the loci: IA=VD/VE−1. To remove the dependency on number of loci, modified statistic rd is used, where var (j, k) are the variance of single loci j and k, respectively:

Exact test for locus differentiation between all pairs of populations was computed by Genepop 3.0 software.22 The threshold of significant differentiation was determined <0.01, more stringent that traditional <0.05 due to relatively small sample size.
Analysis of population structure and assignment of individuals into inferred population clusters was carried out using STRUCTURE version 2.0 software25 (http://pritch.bsd.uchicago.edu). We analyzed three alternative data sets: The data set A (nsamples=1241) consisted of seven Xq13 loci (DXS1066 excluded) for 21 population across from Europe (10), Volga Ural (4) and East-Asia (7). The reference populations of Asian origin included Evenki and Buriats as well as published genotypes from Japanese and four Mongolian populations.17 The data set B (nsamples=889) included eight Xq13 and six Xp22 microsatellites for 10 European, four Volga-Ural and two Siberian populations. The data set C used the same samples as B, but for analyzing only six markers of Xp22. Structure analysis was conducted under linkage model,26 an extension to the original method for inteferring population structure from multilocus data,25 but allowing for linkage between loci. Analysis was conducted with the following parameters: no prior population information, 30 000 burn-in period and 1 000 000 run length. Multiple runs of each data set guaranteed the robustness of the analysis. The number of population clusters was estimated as the value of K that maximized estimated model log-likelihood, log(P(X∣K).
Results
Locus diversities of Xq13 and Xp22 microsatellites
Microsatellites of both studied regions were characterized by locus diversity of similar magnitude, 0.62–0.72 (±s.d. 0.34–0.39) for Xq13 and 0.62–0.73 (±0.35–0.41) for Xp22 (Table 1). Consistent with population genetics theory, reduction in mean number of alleles per locus was found for the Saami and Evenki (genetic drift in small constant populations), as well as Kuusamo (extreme bottleneck) sample. For these populations the reduction in diversity level correlated with lower estimates of population genetics parameter θ: 3.84–5.60 averaged across Xq13 microsatellites and 3.74–5.81 for Xp22 loci compared to 8.42–11.15 (Xq13) and 6.55–12.17 (Xp22) for other populations. In most of the populations, except for the Saami, Evenki, Kuusamo and Mari, every individual carried a unique haplotype constructed of 6 (Xp22) or 8 (Xq13) studied loci.
Background LD between unlinked markers varies among population samples
The ‘baseline’ LD for each population sample was estimated by computing the D′BASE values for all possible pairs (n=48) of unlinked microsatellite loci formed between the Xq13 and Xp22 markers. The mean ‘baseline’ D′BASE varied two times across populations, ranging from 0.166±s.d. 0.052 for Italians to 0.331±s.d. 0.093 in Komi (Figure 1). The median D′BASE follows tightly the mean values indicating the relatively even distribution of the D′BASE values around the mean. However, the maximum D′BASE values equal 1 in several populations, indicating that a pair of unlinked markers can show significant association just by chance. The mean D′BASE was found to be correlated with neither (I) population size – small vs large populations, Kuusamo mean D′BASE=0.289±s.d. 0.099 and the Saami 0.291±0.106 vs Swedes 0.286±0.079; (II) demographic history – constant vs expanded populations, the Saami and Evenki D′BASE=0.291±0.106 and 0.234±0.086 vs Estonians and Russians 0.305±s.d. 0.098 and 0.203±0.078); nor (III) sampling – from one county vs across population, Dutch D′BASE=0.209±0.079 vs Finns 0.191±0.082. Consistently with previous reports,21, 22 we found negative correlation between the mean D′BASE and sample size (Corr. Coef.=−0.911). Our D′ values computed for unlinked microsatellite loci on X chromosome are higher than usually obtained for unlinked SNPs, where D′BASE<0.2.5, 13 The higher baseline D′ values could either result from different marker properties (SNPs vs microsatellites) and/or distinct LD patterns on X-chromosome due to smaller effective population size as well as two times reduced recombination events compared to autosomes. This indicates the importance of estimating the baseline LD for each population sample and marker set used in any particular study aiming to study LD patterns.

Notched boxplot for the distribution of D′ values between 48 unlinked locus pairs (D′BASE) formed between Xq13 and Xp22 microsatellite markers in studied populations. The boxes represent the 25th and 75th percentiles. The median D′ is denoted as the line that bisects the boxes. Notches represent a robust estimate of the uncertainty about the medians for box to box comparison. The whiskers are lines extending from each end of the box covering the extent of the data on 15 X interquartile range. Crosses represent the outlier D′BASE values.
Different patterns of LD across Xp22 and Xq13
Allelic association between microsatellite loci across Xp22 and Xq13 in each population was studied by three statistics: (a) multiallelic extension of Lewontin's metric D′, (b) Fisher's exact test for the significant departure from linkage equilibrium, (c) multilocus association parameter of rd. Xp22 stands out as relatively LD – poor region, most of the populations exhibit 0–2 significant (0.01<P<0.05 from Fisher's exact test) associations for the 15 studied marker-pairs (data not shown). LD across Xp22 exceeds significantly X-chromosomal D′BASE only between closely linked loci for the Saami, Mari and Udmurt, and for markers further apart in Kuusamo and Udmurt sample (Table 2). In the case of the Udmurt, apparently a recent mutation in DXS987 is responsible for creating LD as this marker was involved in three of four associations across Xp22. Consistently, the multilocus LD parameter rd values (<0.05, except the Saami) across 10 Mb Xp22 region refer minimal association of markers (Figure 2).

Comparative multilocus LD values (rd) for the analyzed (1) a 10 kb subregion (five markers, DXS8037 to DXS986) from Xq13 (black columns); (2) 10 kb region (six markers) from Xp22 (white columns) in each studied population, calculated from the distribution of allelic mismatches between pairs of individuals over all loci. LD estimation is based on the variance of the number of pairwise differences among samples that have been subjected to genetic analysis at the multiple loci. Maximum value of rd=1 referring to absolute linkage disequilibrium across the whole region.
In contrast to the LD-poor Xp22 region, Xq13 shows a more diverse picture of the regional LD structure among populations both for overall association parameter rd (Figure 2) as well as for pairwise LD patterns (Figure 3). For all studied populations, Xq13 exhibited stronger LD compared to Xp22 (Figure 2, Table 2). The Saami has the strongest multilocus LD (rd=0.3), the Evenki, Mari, Udmurt and Kuusamo show intermediate values (rd=0.1–0.3) and for the rest of the populations rd<0.1. Also, the results from Mann–Whitney U-test comparing the distribution of D′ values across Xq13 with D′BASE estimates between unlinked markers and thus minimizing sample size effect, demonstrate ‘useful’, background-level exceeding LD for the above-mentioned populations (Table 2). Pairwise D′ estimates for the microsatellites across Xq13 (Figure 3) correlated largely with the calculations for the significance of the association by Fisher's exact test (data not shown). Current extended data of the LD structure across Xq13 reveals that additionally to previously described isolates with distinct demographic histories,10, 11, 12, 17, 27 also the Volga-Ural populations of Mari, Udmurt, Komi and Chuvash harbor increased level of LD across Xq13 compared to other European populations (Figure 3). Consistently, in contrast to single strong association (P<0.05) among 28 studied locus pairs for the majority of populations, Fisher's exact test showed for the Mari 15, for Udmurts 11, for Chuvash 8 and Komi 7 loci in LD. This level of LD is comparable to the LD pattern of Xq13 from Kuusamo isolate (11/28 pairs P<0.05), where the increase in LD levels on X chromosome reflects a recent founder effect. As the current census sizes of these populations (500 000 to 1.8 million) exclude an extreme and recent bottleneck similar to Kuusamo, alternative scenarios could be considered responsible for increased LD.

Patterns of LD for Xq13 microsatellite pairs, estimated as multiallelic D′. Every square represents the mean D′ estimate between pairs of loci from random sampling of 40 individuals from each population sample over 100 replicates. The markers are indicated: 1 – DXS983, 2 – DXS8037, 3 – DXS8092, 4 – DXS1225, 5 – DXS8082, 6 – DXS986, 7 – DXS1066, 8 – DXS995.
European and East Asian populations form two clusters by STRUCTURE analysis: Volga-Ural populations fall to both clusters
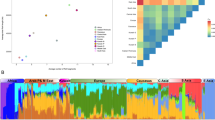
In order to weigh the two alternative demographic scenarios – inner structuring into subgroups or admixture with Asian migrants – responsible for the elevated level of LD in Volga-Ural populations, the genetic structure of the study sample was analyzed by linkage-model based clustering method without prior assignment of individuals into populations.25, 26 Multiple runs for data sets A (nsamples=1241, npop=21, nloci=7) and B (nsamples=889, npop=16, nloci=14) supported the estimate for K=2, indicating two major population clusters among studied samples (Table 3). Data set C (nsamples=889, npop=16, nloci=6) did not resolve the population structure apparently due to low number of markers combined with the unbalanced sampling from East Asia (two populations) compared to Europe (14 populations). Based on data set A, for each individual the proportion of ancestry in both of the clusters was inferred (Figure 4). From the first glance it seems that one of the clusters is enriched in European and the other in Asian populations. Among the studied European populations almost all the individuals were assigned as most probably belonging to the ‘European’ cluster. On the other hand, almost all the Japanese and Evenki belong to the ‘Asian’ cluster. Consistent with known demographic history – admixture with Turkic tribes, as well as admixture LD in these populations shown by Katoh et al17 – the Mongolian populations of Zakhchin, Khoton, Uriankhai exhibit 2/3 of the ‘Asian’ and 1/3 ‘European’ lineages. Minor European contribution was detected also for Buriats and Khalkh. Compared to other Europeans, the Volga-Ural populations of Mari, Chuvash, Udmurt and Komi, have a significant fraction of individuals belonging to the ‘Asian’ cluster. This strongly supports the interpretation of the increase of LD level in these populations owing to admixture with Asian migrants.

Summary of the clustering results (K=2) for the data set A consisting of seven Xq13 microsatellite markers. In 21 studied populations, for each individual (x-axis of each graph) the mean value of the proportion of the ancestry in cluster 1 (white circles) and cluster 2 (gray triangles) was computed (y-axis of each graph). Based on the assignment of the majority of individuals of European origin into cluster 1 and of Asian origin into cluster 2, the two clusters are denoted in the text as ‘European’ and ‘Asian’.
Haplotypes of nonrecombining loci DXS1225-DXS8082 support the hypothesis for admixture LD in Volga-Ural populations
At Xq13, the closest marker pair (162 kb apart) DXS1225–DXS8082 exhibited strong LD (P<0.000001) in all populations, irrespective of population structure or history, studied by us as well as other authors.17, 27 Thus, as we can assume that recombination events between DXS1225 and DXS8082 are extremely rare, new variants are mostly created by mutation in one of the two loci. Table 4 summarizes the frequencies of 13 common haplotypes, present in one or more populations with frequencies exceeding 10%. Total number of DXS1225–DXS8082 haplotypes detected for a population sample ranged 10–27. Number of common haplotypes (>10% frequency) in each studied population ranged from 1 to 5, the haplotype diversities ranged from 0.19 in Japanese and the Saami to 0.44 in a Mongolian tribe of Uriankhai.
At first glance, there is a clear difference between European and Asian haplotype distributions. In European populations, except the Saami, across the vast area from West-Europe to the Urals one major haplotype (210–219) predominates (17 – 40%). Furthermore, in several populations this haplotype extends to neighboring DXS986 across 1.215 Mb. In East Asia, this haplotype is present at low frequency (≤10%). On the contrary, the common haplotype detected in Asia (202–217) ranging from 16% in Uriankhai to 34% in Japanese, is almost absent in Europe. However, the exception here is the Volga-Ural region, where this East Asian haplotype is quite common among Udmurts (15%), Chuvash (10%) and Komi (11%). Overall, the distribution of common haplotypes of DXS1225–DXS8082 in Volga-Ural region is more complex compared to the vast area of the rest of Europe. Consistent with the STRUCTURE results it suggests admixture of mainly European gene pool with East Asians combined with influence of genetic drift. Notably, the allelic distributions of single microsatellites or haplotypes of weakly associated markers are mostly shared between the Volga-Ural and European populations, and have not preserved a footprint of East Asian migration (data not shown).
Comparing DXS1225–DXS8082 haplotype distribution in Finns and the Kuusamo isolate provides vivid evidence for the impact of genetic drift in changing the allele and haplotype frequencies within a short period of time. Although the isolate shares the major haplotype with Finns (28 vs 31%), there are two other enriched haplotypes (both 15%). As one of them, 212–219, is also common (20%) among the Saami living originally at the Kuusamo area, and rare in the rest of Europe, it could also reflect admixture with local Saami people during the establishment of the population.
Discussion
Demographic history has the strongest impact on LD patterns of recombination-poor regions
This study shows that demographic history has strong impact on local LD and haplotype patterns across a 20 Mb, but only 4.74 cM, genomic segment at Xq13. No such strong LD generating effect was detected across a 9.207 Mb region at Xp22 corresponding to 12.01 cM. As microsatellites genotyped for Xq13 and Xp22 were characterized by similar locus diversity, mean number of alleles and population diversity parameter θ, we can leave aside a scenario of higher mutation rate of Xp22 markers responsible for diversifying haplotype and LD patterns. Our result is concordant with the recent simulation study by Stumpf and Goldstein,28 which demonstrated that following the LD-generating event, the differences among genomic regions in preserving a block-like structure depend on recombination rate. In our data set outbred and expanded populations, independent of their size, exhibited significant LD at Xq13 between only one single locus pair DXS1225–DXS8082. The reason for the strong LD between these markers, located 162 kb apart and apparently within an LD desert, is still to be studied. In our data set LD-generating events across Xq13 included not only genetic drift in a small population or severe founder-effect, but also admixture with genetically different migrants. Volga-Ural populations, distributed at the geographic borderline of Europe and Asia, have apparently historically lived in close contact with their East Asian neighbors. Both, population structure and haplotype analysis supported the hypothesis that the increased level of LD in these populations is due to admixture of mostly European genepool with East Asians. This level of LD is similar to the extent observed for X-chromosomal loci in a Bantu-Semidic hybrid population of Lemba.29 For Lemba, similar to Komi and Chuvash, the elevated background LD was observed also for unlinked markers on X-chromosome. The strongest LD across Xq13 region was detected for Udmurt and Mari. There the inner structuring of the population could additionally contribute to the creation of nonrandom allelic associations.
Implications for mapping using LD
Two recent extensive scans for the landscape of LD and haplotype variation across human genome in distant population groups point out that there is a lot of heterogeneity in the LD map as well as haplotype frequencies among populations.30, 31 There is also evidence that the intervals across which LD is detectable depend on marker properties. Varilo et al13 showed that single informative microsatellites provide more power to detect long-range LD than did single SNPs or even 3–5 SNP haplotypes, It has been shown that long-range microsatellite data can be used to predict short-range LD between SNPs and thus assist in initial association analysis.32, 33 In addition, our study emphasizes the importance of calculating the baseline LD between unlinked markers for each data set used in the study. The true indication of the increase of LD in a particular genomic region is in comparison to the baseline LD.
Data on X-chromosomal microsatellites show that the footprint of a demographic event persists longer in haplotype distribution within a region of low crossing-over rate than in the information content of a single marker or between the actively recombining markers. The distribution of the haplotypes of strongly associated DXS1225–DXS8082 markers varies between populations, memorizing the demographic events of a population. For example, when bottleneck is accompanied with low level admixture, the few migrant haplotypes might drift frequent in the descendant population as proved by the 212–219 haplotype distribution in the Kuusamo and Saami (Table 4). Also, inner structuring within populations can lead to additional haplotype frequency differences. There are more and more data, which highlight the importance of taking into account the particular population history and its impact on regional LD patterns. Laan and Pääbo34 have compared the allelic associations around renin–binding protein RnBP, a component of the renin–angiotensin system, in the Saami and Finns. The minor allele of an SNP within the gene, T61C, present both on the Saami (21%) as well as Finns (19%) as common mutation, was associated with different alleles and haplotypes of flanking microsatellites in two populations. The recently described association between SNPs of the CARD15 (NOD2) gene and Crohn's disease35 did not find any support in the respective study with Korean patients.36 The three disease-associated SNPs sharing a common haplotypic background were absent in the Korean sample and the LD pattern across CARD15 differed between the studied populations.
In conclusion, our study demonstrates that demographic events leave their prolonged imprint on LD patterns across recombination-poor genomic regions. Consequently, as the haplotype distribution within LD-rich blocks might exhibit much more variability among populations than previously expected, the key for successful gene mapping studies is detailed understanding of the history, structure and variation of the study population.
References
Kerem B, Rommens JM, Buchanan JA et al.: Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245: 1073–1080.
Hästbacka J, de la Chapelle A, Kaitila I et al. Nat Genet 1992; 2: 204–211.
Risch N, Merikangas K : The future of genetic studies of complex human traits. Science 1996; 273: 1516–1517.
Kruglyak L : Prospects of whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 1999; 22: 139–144.
Ardlie KG, Kruglyak L, Seielstad M : Patterns of linkage disequilibrium in the human genome. Nat Rev Genet 2002; 3: 299–309.
Jeffreys AJ, Kauppi L, Neumann R : Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet 2001; 29: 217–222.
Couzin J : New mapping project splits the community. Science 2002; 296: 1392–1393.
Wall JD, Pritchard JK : Haplotype blocks and linkage disequilibrium in the human genome. Nat Rev Genet 2003; 4: 587–597.
Schaffner SF : The X chromosome in population genetics. Nat Rev Genet 2004; 5: 43–51.
Laan M, Pääbo S : Demographic history and linkage disequilibrium in human populations. Nat Genet 1997; 17: 435–438.
Kaessmann H, Zöllner S, Gustafsson A et al.: Extensive linkage disequilibrium in small human populations in Eurasia. Am J Hum Genet 2002; 70: 673–685.
Varilo, T, Laan M, Hovatta I, Wiebe V, Terwilliger JD, Peltonen L : Linkage disequilibrium in isolated populations: Finland and a young subpopulation of Kuusamo. Eur J Hum Genet 2000; 8: 604–612.
Varilo T, Paunio T, Parker A et al.: The interval of linkage disequilibrium (LD) detected with microsatellite and SNP markers in chromosomes of Finnish populations with different histories. Hum Mol Genet 2003; 12: 51–59.
Khusnutdinova E : Molecular ethnogenetics of the populations of Volga-Ural region. Ufa: Gilem Press, 1999, Russia (in Russian).
Bermisheva M, Tambets K, Villems R, Khusnutdinova E : Diversity of mitochondrial DNA haplogroups in ethnic populations of the Volga-Ural region. Mol Biol (Moskow) 2002; 36: 990–1001, (in Russian).
Laakso J : Uralilaiset Kansat; WSOY, Porvoo-Helsinki-Juva,: 1991, Finland (in Finnish).
Katoh T, Mano S, Ikuta T et al.: Genetic isolates in East Asia: a study of linkage disequilibrium in the X-chromosome. Am J Hum Genet 2002; 71: 395–400.
Schneider S, Roessli D, Excoffier L : Arlequin ver. 2.000: A software for population genetic data analysis.; Genetics and Biometry Laboratory 2000, University of Geneva, Switzerland.
Nielsen R : A maximum likelihood approach to population samples of microsatellite alleles. Genetics 1997; 146: 711–716.
Abecasis GR, Cookson WO : GOLD – graphical overview of linkage disequilibrium. Bioinformatics 2000; 16: 182–183.
Weiss KM, Clark AG : Linkage disequilibrium and the mapping of complex traits. Trends Genet 2002; 18: 19–24.
Tenesa A, Wright AF, Knott SA et al.: Extent of linkage disequilibrium in a Sardinian sub-isolate: sampling and methodological considerations. Hum Mol Genet 2004; 13: 25–33.
Raymond M, Rousset F : GENEPOP ver 1.2.: Population genetics software for exact tests and ecumenicism. J Hered 1995; 86: 248–249.
Brown AHD, Feldman MW, Nevo E : Multilocus structure of natural populations of Hordeum spontaneum. Genetics 1980; 96: 523–536.
Pritchard JK, Stephens M, Donnelly P : Inference of population structure using multilocus genotype data. Genetics 2000; 155: 945–959.
Falush D, Stephens M, Pritchard JK : Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 2003; 164: 1567–1587.
Angius A, Bebbere D, Petretto E et al.: Not all isolates are equal: linkage disequilibrium analysis on Xq13.3 reveals different patterns in Sardinian sub-populations. Hum Genet 2002; 111: 9–15.
Stumpf MPH, Goldstein DB : Demography, recombination hotspot intensity, and the block structure of linkage disequilibrium. Curr Biol 2003; 13: 1–8.
Wilson JF, Goldstein DB : Consistent long-range linkage disequilibrium generated by admixture in a Bantu-Semitic hybrid population. Am J Hum Genet 2000; 67: 926–935.
Clark AG, Nielsen R, Signorovitch J et al.: Linkage disequilibrium and interference of ancestral recombination in 538 single-nucleotide polymorphism clusters across the human genome. Am J Hum Genet 2003; 73: 285–300.
Salisbury BA, Pungliya M, Choi JY, Jiang R, Sun XJ, Stephens JC : SNP and haplotype variation in the human genome. Mutation Res 2003; 526: 53–61.
Schulze T, Chen Y-S, Akula N et al.: Can long range microsatellite data be used to predict short-range linkage disequilibrium? Hum Mol Genet 2002; 11: 1363–1372.
Gretarsdottir S, Thorleifsson G, Reynisdottir ST et al.: The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet 2003; 35: 131–138.
Laan M, Pääbo S : Mapping genes by drift-generated linkage disequilibrium. Am J Hum Genet 1998; 63: 654–656.
Hugot J-P, Chamaillard M, Zouali H et al.: Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 2001; 411: 599–603.
Croucher PJP, Mascheretti S, Hampe J et al.: Haplotype structure and association to Crohn's disease of CARD15 mutations in two ethnically divergent populations. Eur J Hum Genet 2003; 11: 6–16.
Acknowledgements
We thank Drs Lars Beckman, Cristian Capelli, Andreas Kindmark, Peter de Knijff, Leena Peltonen, Mikchail Voevoda, and G Weichhold for DNA samples. Dr Tatjana Victorova is specifically acknowledged for assistance with collecting the Volga-Ural samples. Dr Molly Przeworski is acknowledged for methodological advise, discussions and comments on the early version of the manusctript. Dr Kalle Olli is thanked for statistical help, Lauri Kaplinski for assistance in preparation Figure 3 and Dr Roger Horton for editing the English text. ML has been supported by the Alexander-von-Humboldt Stiftung and Wellcome Trust International Senior Research Fellowship in Biomedical Science in Central Europe. MR is supported by Core Grant No. 0182649s04 of the Estonian Ministry of Education and Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Laan, M., Wiebe, V., Khusnutdinova, E. et al. X-chromosome as a marker for population history: linkage disequilibrium and haplotype study in Eurasian populations. Eur J Hum Genet 13, 452–462 (2005). https://doi.org/10.1038/sj.ejhg.5201340
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201340
Keywords
This article is cited by
-
Development of a new screening method for faster kinship analyses in mass disasters: a proof of concept study
Scientific Reports (2022)
-
X-chromosomal STR-based genetic structure of Sichuan Tibetan minority ethnicity group and its relationships to various groups
International Journal of Legal Medicine (2018)
-
Simple strategies for haplotype analysis of the X chromosome with application to age-related macular degeneration
European Journal of Human Genetics (2011)
-
Validation of six closely linked STRs located in the chromosome X centromere region
International Journal of Legal Medicine (2010)
-
An INDEL polymorphism at the X-STR GATA172D05 flanking region
International Journal of Legal Medicine (2009)
