
Abstract
Mutations in the CLCN1 gene, encoding a muscle-specific chloride channel, can cause either recessive or dominant myotonia congenita (MC). The recessive form, Becker's myotonia, is believed to be caused by two loss-of-function mutations, whereas the dominant form, Thomsen's myotonia, is assumed to be a consequence of a dominant-negative effect. However, a subset of CLCN1 mutations can cause both recessive and dominant MC. We have identified two recessive and two dominant MC families segregating the common R894X mutation. Real-time quantitative RT-PCR did not reveal any obvious association between the total CLCN1 mRNA level in muscle and the mode of inheritance, but the dominant family with the most severe phenotype expressed twice the expected amount of the R894X mRNA allele. Variation in allelic expression has not previously been described for CLCN1, and our finding suggests that allelic variation may be an important modifier of disease progression in myotonia congenita.
Similar content being viewed by others

Introduction
Myotonia congenita, MC (MIM 118425) is an inherited disorder characterized by delayed skeletal muscle relaxation caused by a diminished activity of the voltage-gated chloride channel ClC-1 in the sarcolemma. Traditionally, the disease has been divided into Thomsen's myotonia (MIM160800), which is inherited autosomal dominantly and Becker's myotonia (MIM255700), which is inherited autosomal recessively. The mode of inheritance is usually causative for the clinical classification as Thomsen or Becker's myotonia. Both conditions usually have onset during childhood and are characterized by prominent myotonia that is reduced by continued muscle activity, the so-called ‘warm up’ effect. Patients with the recessive form are generally more severely affected with occasional occurrence of permanent muscle weakness and atrophy. The dominant and recessive forms are due to allelic mutations in the CLCN1 gene located on 7q35, and since its discovery in 19911, 2 more than 65 different mutations have been described.3, 4, 5 The vast majority of these are found in recessive pedigrees, whereas approximately only 10 mutations exclusively result in dominant MC presumably through a dominant-negative effect.6 More that 10 mutations have been associated with both recessive and dominant MC,3 making a clear distinction between the two modes of inheritance of the disease difficult. This peculiar phenomenon has been explained by reduced penetrance of dominant-negative mutations, incomplete dominance, founder effect and incomplete mutation detection.7, 8, 9, 10 The obscured manifestation of CLCN1 defects is further complicated by the observation that some families harbor three CLCN1 mutations11, 12 and atypical cases where myotonia only appears during pregnancies.13 The Clc-1 channel is a homodimer and dominant MC is thought to result from dominant-negative mutations poisoning the complex, whereas recessive MC presumably originates from a loss-of-function of the channel.
Several mutations have been characterized by in vitro patch-clamp assays, but even mutations with electrophysiologically characteristics consistent with dominant inheritance have been found in recessive pedigrees as well.14, 15 R894X is one of the most common CLCN1 mutations present in many different populations, and is the prime mutation for which both recessive and dominant inheritance has been reported. The mutation is located in the last exon of the CLCN1 gene and leads to a truncation of the terminal 94 amino acids.
We have previously identified four independent families harboring the common R894X mutation16 and present here mRNA analysis, isolated from patients' muscle biopsies, demonstrating that the CLCN1 gene is subject to variation in allelic expression, which may be a component of disease progression in myotonia congenita.
Materials and methods
Human subjects
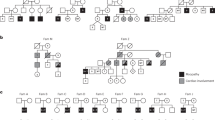
The human subjects for this study were submitted as part of a genetic testing service. After approval from the local ethics committee and receipt of informed consent, selected family members were asked to have a muscle needle biopsy from the M. vastus lateralis performed for analysis of the CLCN1 mRNA expression level. Five individuals volunteered for biopsy (Figure 1), which was undertaken upon receipt of written informed consent.

Pedigrees of the four families segregating the R894X mutation. (a) and (b) depict the recessive pedigrees, whereas (c) and (d) show the dominant R894X segregation. The thick arrow identifies the proband whose entire CLCN1 gene was sequenced, whereas other family members were tested only for identified mutation(s)16 and their CLCN1 genotype is indicated. The thin arrow identifies individuals from whom a muscle biopsy was obtained. Filled symbols denote the presence of a myotonia.
Genotyping
Initial genotyping has been described elsewhere.16 In summary, genomic DNA from the probands was extracted according to standard methods and all 23 exons of CLCN1 were amplified by polymerase chain reaction (PCR) followed by sequencing using intronic primers as described by Kubisch et al. 14 Identified mutations were subsequently confirmed in a new PCR and sequencing reaction. The CLCN1 sequence from the two probands, who also volunteered for cDNA analysis, did not show any differences in the primer sites for mRNA analysis.
mRNA isolation and quantification
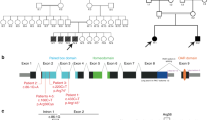
Four of the five biopsies were divided in two and processed independently. Total mRNA was extracted using TRIzol (Invitrogen, Taastrup, Denmark) and cDNA was generated using Superscript II™ (Invitrogen) in combination with a dT16 primer, both according to the manufacturer's description. The CLCN1 cDNA was evaluated by sequencing PCR products from two overlapping fragments of the N- and C- terminal part of the transcript using the primers described by Grunnet et al.5 A real-time semiquantitative PCR analysis was performed on an SDS7000 Sequence Detection System using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, USA). Briefly, cDNA-specific primers for CLCN1 and the reference gene GUSB were designed according to the PrimerExpress software (CLCN1-exon21–22 AAGAAAACAACCCAG/GATTCC; TCAGGTGACATGTTATCCACTAAATCT, GUSB-exon5–6 TCATTTGGAATTTTGCCGATT; GCACTCTCGTCGGTG/ACTGTT, vertical bar indicates exon–exon junction). The two PCR reactions were evaluated individually on a 5 × dilution series for the generation of standard curves. As demonstrated in Figure 2, the two PCR reactions exhibited similar amplification efficiencies, allowing the raw data to be analyzed by the comparative CT methods (described in the User Bulletin 2 from Applied Biosystems).

Real-time quantitative RT-PCR of CLCN1 and the reference gene GSUB. (I) and (II) show the raw data (5 × dilution series) from which the standard curves were generated. All dilutions were run in duplex and the curves were generated in three independent experiments. Both PCRs were specific for cDNA and exhibited similar amplification efficiencies as indicated by the slope of the standard curves. (III) Relative expression of CLCN1 normalized to family A. The vertical bars denote 2 SD generated from four independent experiments with a minimum of two assays per biopsy. A, B, C, and D correspond to the families in Figure 1. The difference between families A and B was highly significant (P=0.0001, unpaired Student's t-test), whereas there were no significant differences between families A and C, and B and C.
Allelic quantification
The R894X mutation disrupts a TaqI restriction site, which was used to distinguish wild-type from mutant mRNA alleles. A cDNA-specific PCR reaction was designed (Fwd fam-CTCGCTTACGTGACCAGCATG; Rvs CTCAGGCAGGTTCCAGTTCTCT) giving rise to a 189 bp fluorochrome-labeled PCR product that could be monitored on an ABI 310 Genetic Analyzer (Applied Biosystems, Foster City, USA). A homozygous wild-type product yields a 136 bp fluorochrome-labeled fragment after TaqI restriction, whereas a homozygous mutant product is resistant to TaqI restriction. A wild-type and mutant sequence was cloned into the pGEM-T vector (Promega, Madison, USA) and purified PCR fragments were mixed in different ratios, heated to 95°C and slowly cooled to room temperature (heteroduplex formation), TaqI restricted and analyzed on the ABI310 using the GeneScan software. The proportions of the individual peak areas were calculated and a standard curve was generated, which revealed a convincing relation between the different peak areas and the original mutant/wild-type ratio (Figure 3 II). To further evaluate any possible effect of heteroduplex formation, the quantification assay was initially performed on identical PCR reactions terminated after 18, 20, 22, 24, 26, 28 and 30 PCR cycles. No obvious difference in the relation of wild-type and mutant allele could be detected across the samples, and the assay was thereafter monitored after 30 PCR cycles.

Relative quantification of the wild type and mutant mRNA alleles. (I) The 189 bp peak corresponds to the R894X allele, whereas the 136 bp peak corresponds to the wild-type allele (smaller peaks are size standards). The upper and lower panels show the allele quantification of the individuals C and D, respectively. (II) Different amounts of cloned mutant and wild-type fragments were used to generate a standard curve validating the assay. Vertical bars represent one standard deviation generated from three independent quantifications. Possible hetero-duplexes formation did not seem to interfere with the quantification (materials and methods). (III) Relative amount of the R894X allele. Vertical bars indicate 1 SD generated from five independent assays. No sequence difference was found in the primer sites between the probands in families A and D, excluding allelic PCR bias.
Results
Clinical evaluation
We have previously identified four unrelated families segregating the R894X mutation.16 In two families (Figure 1a and b), the mutation segregated as recessive with heterozygous family members having neither clinical nor electromyographic signs of myotonia, whereas in the two other families (C and D), the mode of inheritance was dominant. Family C had an atypical late onset (20–30 years of age), which may be regarded as an unusually mild phenotypic expression. In family D, the age of onset was 7 years, which is comparable to the age of onset in most dominant cases of myotonia congenita. Further, in patients from family D, the compound muscle action potential evoked from a hand muscle showed significant decrement on 10 Hz repetitive stimulation, whereas patients from family C had borderline decrement or none at all, again suggesting a less severe phenotype than that found in family D.
mRNA expression
We speculated whether the level of CLCN1 mRNA expression reflected the dual mode of inheritance. One individual each from families A–C, and two from family D volunteered for muscle biopsy for mRNA analysis (Figure 1). Initial cDNA analysis did not reveal the presence of any abundant alternative spliced transcripts. Both mutations in the proband from family A could be detected on the mRNA level (not shown). The relative expression level of CLCN1 was determined in four independent experiments and related to the recessive family A (Figure 2). Although the expression level varied almost a factor 2 between the highest and lowest expression, there was no obvious relation to the mode of inheritance. The level of expression in the individual from family B, who was asymptomatic, did not differ significantly from the CLCN1 level in the dominant family C (P=0.14) but was lower compared to family D (P<0.012). The age of the patients at biopsy ranged from 34 years (individual C) to 65 years (individual B) and there was no striking relation between the relative CLCN1 mRNA level and age as suggested.17 Individual B was, however, the oldest, whereas the individuals from families A and D were between 49 and 52 years of age.
Allelic quantification
The variation in expression observed between the different families prompted us to speculate whether a difference in allelic expression, rather than the total mRNA level per se was the underlying cause for the dual mode of inheritance. An assay was developed allowing discrimination of the wild-type and R894X mRNA allele and the evaluated assay was consequently used to quantify the relative amount of the R894X cDNA in the different cDNA preparations. Surprisingly, both individuals from the dominant family D expressed almost twice as much R894X mRNA relative to the wild-type allele, compared to the other families (Figure 3III). There appeared to be no gross difference between the asymptomatic individual in the recessive family B, and the individuals from families A and C. Ideally, we expected a 1:1 ratio of the two alleles. However, the mutant allele in families A–C accounted for only 35–39% of total mRNA.
Discussion
The existence of a single mutation that in some families exhibits a dominant phenotype, whereas in other families behaves recessive, is a most peculiar genetic phenomenon. It is well established that the R894X mutation in the CLCN1 gene is an example of such a mutation. Based on a functional characterization in Xenopus oocytes, Meyer-Kleine et al.18 suggested that the mutation conferred electrophysiological properties intermediate between pure recessive and dominant mutations, and that coexpression with wild-type CLCN1 yielded electrophysiological results close to normal. We therefore analyzed whether the level of CLCN1 mRNA expression was decisive for the dual mode of inheritance. The highest and lowest CLCN1 mRNA levels found in the recessive families A and B, respectively, suggest that the expression level of CLCN1 is not a key factor in distinguishing recessive from dominant MC. However, we cannot exclude the possibility that expression level variation may have a minor influence on the phenotypic variability. The G285E mutation from the proband in family A could be detected on cDNA indicating correct mRNA maturation. Previous reports have suggested that up to 27% of CLCN1 cDNA is subject to alternative spliced forms that results in the presence of premature termination codons.19, 20 These transcripts are most likely rapidly eliminated through nonsense-mediated decay and escape detection by our cDNA analysis. In this respect, it is important to note that the two PCR fragments used for CLCN1 mRNA and allele quantification, respectively, are located close to each other in the far C-terminal part of the transcript. It is therefore unlikely that the potential alternatively spliced forms have a significantly impact on the analysis.
ClC-1 channels are known to form homodimers,21 and recessive MC is believed to result from two loss-of-function mutations, whereas dominant MC mutations are presumed to exhibit a dominant-negative effect, ‘poisoning’ the channel. Therefore, the variation in expression observed between the different families prompted us to investigate whether a difference in allelic expression, rather than the total mRNA level per se was the underlying cause for the dual mode of inheritance.
Our allelic quantification assay clearly demonstrate that the CLCN1 gene is subject to variation in allelic expression, and that the dominant severely affected family D expressed twice the amount of R894X mRNA. Such allelic variation has just recently been identified in other genes 22 and implicated as disease-causing.23 In fact, allelic variation may be a quite common phenomenon 24 and is likely to be a general mechanism contributing to human phenotypic diversity.25, 26 The specific mechanism behind the difference in allelic expression is presently unknown but could be based on polymorphisms at the promoter or enhancer regions. Alternatively, the global chromatin organization of the CLCN1 locus could possibly facilitate allelic expression variation as well as differences in the RNA processing machinery.
The limited number of subjects investigated makes it difficult to draw any firm conclusions about exactly to which extent the difference in allelic expression contribute to the distinction between dominant and recessive myotonia congenita. Ideally, more individuals should be tested; this is, however, extremely difficult due to the very low occurrence of affected individuals. Nonetheless, our study shows that the two alleles of the CLCN1 gene are differentially expressed.
If the difference in allelic expression is a key determinant for dominant versus recessive inheritance of the R894X mutation, it is surprising that family C had an R894X mRNA expression comparable to that of recessive patients. However, the two dominant families exhibited distinct clinical phenotypes with late onset of the disease in family C and electrophysiological findings suggestive of a less severe phenotype than that of family D.
Although more individuals should ideally be tested, our findings raise the question whether variation in allelic expression is a component in determining the phenotype of congenital myotonia. In a study of nine recessive families segregating the R894X mutation, Sun et al.27 identified three carrier individuals from different families, who all exhibited latent myotonia. Thus, even within the same family carriers of the recessive R894X mutation can exhibit distinct clinical phenotypes. Whether this is related to variation in allelic expression is currently unknown.
Shifting the proportion of transcripts towards the mutant allele might result in a greater quantity of ‘poisoned’ ClC-1 channels that could give rise to the dominant phenotype. The underlying mechanism for this shift is not known, but the existence of high and low expressed alleles offers an attractive explanation for the observed inconsistency in the mode of inheritance for some of the CLCN1 mutations.
Variation in allelic expression would also provide the explanation why other mutations in the CLCN1 gene can behave both dominant and recessive. For instance, the F307S mutation was initially found in a dominant family, and electrophysiological characterization supported the dominant nature of the mutation.14 However, the same mutation has subsequently been found the recessive families.3 It is quite possible that allelic variation in favor for the mutation is recognized as a dominant phenotype, whereas a shift to the wild-type allele leads to recessive inheritance. Thus, expression of the normal allele below a critical threshold could result in altered disease progression. Such a scenario has just recently been suggested for the etiology of Marfan syndrome.28
It is interesting to note that a single mutation in the SOD1 gene likewise can result in either recessive or dominant amyotrophic lateral sclerosis, and that all recessive families share the same haplotype whereas dominant families do not.29 Thus, this may represent another example of the influence of differential allelic expression on disease progression. Mutations in another chloride channel gene, CLCN7, can result in both dominant and recessive osteopetrosis,30 but as for the SOD1 gene mutation, it remains to be tested whether this dual pattern of inheritances is a result of differential allelic expression.
References
Steinmeyer K, Ortland C, Jentsch TJ : Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature 1991; 354: 301–304.
Koch MC, Steinmeyer K, Lorenz C et al: The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 1992; 257: 797–800.
Pusch M : Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum Mutat 2002; 19: 423–434.
Wu FF, Ryan A, Devaney J et al: Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain 2002; 125: 2392–2407.
Grunnet M, Jespersen T, Colding-Jørgensen E et al: Characterization of two new dominant clc-1 channel mutations associated with myotonia. Muscle Nerve 2003; 28: 722–732.
Steinmeyer K, Lorenz C, Pusch M, Koch MC, Jentsch TJ : Multimeric structure of ClC-1 chloride channel revealed by mutations in dominant myotonia congenita (Thomsen). EMBO 1994; 13: 737–743.
Mailander V, Heine R, Deymeer F, Lehmann-Horn F : Novel muscle chloride channel mutations and their effects on heterozygous carriers. Am J Hum Genet 1996; 58: 317–324.
Koty PP, Pegoraro E, Hobson G et al: Myotonia and the muscle chloride channel: dominant mutations show variable penetrance and founder effect. Neurology 1996; 47: 963–968.
Zhang J, George Jr AL, Griggs RC et al: Mutations in the human skeletal muscle chloride channel gene (CLCN1) associated with dominant and recessive myotonia congenita. Neurology 1996; 47: 993–998.
Plassart-Schiess E, Gervais A, Eymard B et al: Novel muscle chloride channel (CLCN1) mutations in myotonia congenita with various modes of inheritance including incomplete dominance and penetrance. Neurology 1998; 50: 1176–1179.
Sloan-Brown K, George Jr AL : Inheritance of three distinct muscle chloride channel gene (CLCN1) mutations in a single recessive myotonia congenita family. Neurology 1997; 48: 542–543.
Papponen H, Toppinen T, Baumann P et al: Founder mutations and the high prevalence of myotonia congenita in northern Finland. Neurology 1999; 53: 297–302.
Lacomis D, Gonzales JT, Giuliani MJ : Fluctuating clinical myotonia and weakness from Thomsen's disease occurring only during pregnancies. Clin Neurol Neurosurg 1999; 101: 133–136.
Kubisch C, Schmidt-Rose T, Fontaine B, Bretag AH, Jentsch TJ : ClC-1 chloride channel mutations in myotonia congenita: variable penetrance of mutations shifting the voltage dependence. Hum Mol Genet 1998; 7: 1753–1760.
Jurkat-Rott K, Lehmann-Horn F, Rüdel R : Chloride and sodium channel myotonias; in Rose MR, Griggs RC (eds): Channelopathies of the nervous system. Boston, MA: Butterworth-Heinemann; 2000, pp 141–157.
Colding-Jørgensen E, Dunø M, Schwartz M, Vissing J : Decrement of compound muscle action potential is related to mutation type in myotonia congenita. Muscle Nerve 2003; 27: 449–455.
Pierno S, De Luca A, Beck CL et al: Aging-associated down-regulation of ClC-1 expression in skeletal muscle: phenotypic-independent relation to the decrease of chloride conductance. FEBS Lett 1999; 449: 12–16.
Meyer-Kleine C, Steinmeyer K, Ricker K, Jentsch TJ, Koch MC : Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am J Hum Genet 1995; 57: 1325–1334.
Charlet-B. N, Savkur RS, Gopal S et al: Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Cell 2002; 10: 45–53.
Mankodi A, Urbinati CR, Yuan QP et al: Expanded CUG repeats trigger aberrant splicing of CLC-1 chloride channel pre-mRNA and hyperexcitability of the skeletal muscle in myotonic dystrophy. Cell 2002; 10: 35–44.
Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R : X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 2002; 415: 287–294.
Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW : Allelic variation in human gene expression. Science 2002; 297: 1143.
Bray NJ, Buckland PR, Williams NM et al: A haplotype implicated in schizophrenia susceptibility is associated with reduced COMT expression in human brain. Am J Hum Genet 2003; 73: 152–161.
Lo HS, Wang Z, Hu Y et al: Allelic variation in gene expression is common in the human genome. Genome Res 2003; 13: 1855–1862.
Cheung VG, Conlin LK, Weber TM et al: Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet 2003; 33: 422–425.
Cowles CR, Joel NH, Altshuler D, Lander ES : Detection of regulatory variation in mouse genes. Nat Genet 2002; 32: 432–437.
Sun C, Tranebjaerg L, Torbergsen T et al: Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia. Eur J Hum Genet 2001; 9: 903–909.
Hutchinson S, Furger A, Halliday D et al: Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet 2003; 12: 2269–2276.
Al-Chalabi A, Andersen PM, Chioza B et al: Recessive amyotrophic lateral sclerosis families with the D90A SOD1 mutation share a common founder: evidence for a linked protective factor. Hum Mol Genet 1998; 7: 2045–2050.
Cleiren E, Benichou O, Van Hul E et al: Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the CLCN7 chloride channel gene. Hum Mol Genet 2001; 10: 2861–2867.
Acknowledgements
We are thankful to the members of the families for their participation in this study. Dr Søren Nørby is thanked for critically reading the manuscript. All investigations were carried out in concordance with Danish law.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dunø, M., Colding-Jørgensen, E., Grunnet, M. et al. Difference in allelic expression of the CLCN1 gene and the possible influence on the myotonia congenita phenotype. Eur J Hum Genet 12, 738–743 (2004). https://doi.org/10.1038/sj.ejhg.5201218
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201218
Keywords
This article is cited by
-
Skeletal muscle ClC-1 chloride channels in health and diseases
Pflügers Archiv - European Journal of Physiology (2020)
-
A large intragenic deletion in the CLCN1 gene causes Hereditary Myotonia in pigs
Scientific Reports (2019)
-
Becker’s myotonia: novel mutations and clinical variability in patients born to consanguineous parents
Acta Neurologica Belgica (2018)
-
Clinical, Molecular, and Functional Characterization of CLCN1 Mutations in Three Families with Recessive Myotonia Congenita
NeuroMolecular Medicine (2015)
-
In vitro analysis of splice site mutations in the CLCN1 gene using the minigene assay
Molecular Biology Reports (2014)
