
Abstract
The Rapp–Hodgkin syndrome (RHS, MIM 129400) corresponds to a rare form of anhydrotic ectodermal dysplasia, which shares some features with the ectrodactyly, ectodermal dysplasia and cleft lip/palate syndrome (EEC, MIM 604292) resulting from TP63 mutations. We report here, in two unrelated patients with RHS, the identification of two distinct TP63 mutations, corresponding to a novel frameshift mutation (1709DelA, exon 14) located downstream the sterile alpha motif (SAM) domain and to a missense mutation (R279H, exon 7) within the DNA binding domain. Functional analysis of the R279H mutation, which had previously been reported in several EEC families, shows that this mutation disrupted the dominant negative activity of the ΔNp63α and γ isoforms on the transcriptional activity of TP53. This report shows, on a molecular basis, that RHS is also an EEC-like syndrome resulting from mutations of the TP63 gene, and highlights the wide phenotypic spectrum associated to TP63 mutations.
Similar content being viewed by others
Introduction
The Rapp–Hodgkin syndrome (RHS, MIM 129400) corresponds to a rare form of anhydrotic ectodermal dysplasia.1 The RHS phenotype variably includes cleft lip/palate, small mouth, narrow nose, coarse and wiry hairs progressing to alopecia in adults, oligodontia or anodontia, hypoplasia of the nails, abnormalities of the lacrimal ducts, deformed ears and ear canals and diverse genitourinary abnormalities especially hypospadias in males.2,3,4,5,6,7,8,9
The fact that RHS corresponds to a specific clinical entity is controversial: indeed, Cambiaghi et al10 have suggested that RHS and ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (AEC or Hay–Wells syndrome, MIM 106260) could correspond to the same entity. The overlapping phenotype of RHS with the ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome (EEC, MIM 604292) and the association, within the same family, of RHS and EEC9 have suggested that RHS was a phenotypic variation of the EEC syndrome.
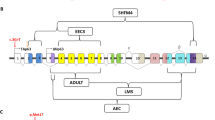
The EEC syndrome was shown to result from mutations of the TP63 gene located on 3q27,11 a homolog of the TP53 tumor suppressor gene (Figure 1). TP63 mutations were subsequently identified in several other autosomal disorders sharing some features with EEC: AEC,12 limb–mammary syndrome (LMS, MIM 603543),13 acro-dermato-ungual-lacrimal-tooth syndrome (ADULT, MIM 103285)14 and nonsyndromic split-hand/foot malformation (SHFM, MIM 605289).15

Schematic representation of TP63. (a) Genomic organization of the TP63 gene showing (i) the two transcriptional start sites, which give rise to TP63 isoforms with (TAp63) or without (ΔNp63) the transactivation domain, and (ii) the various splicing leading to the α, β and γ isoforms. Black boxes represent untranslated regions. Symbols within exons correspond to the different functional domains. The positions of the R279H and 1709DelA mutations, detected in the two patients with RHS, are indicated. (b) The different conserved functional protein domains are indicated. SAM: sterile alpha motif domain, TID: transactivation inhibitory domain.
We report here in two unrelated patients with RHS the identification of two distinct TP63 mutations, demonstrating, on a molecular basis, that RHS is also an EEC-like syndrome.
Material and methods
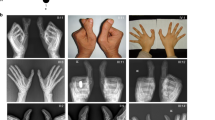
Patient 1, a 32-year-old male born from nonconsanguineous healthy parents, was referred to us for genetic counselling in the context of an hypohydrotic ectodermal dysplasia (Table 1). He presented cleft palate, diffuse hypotrichosis with short dystrophic and thick hair, sparse eyelashes and eyebrows, mild hypohydrosis and partial anodontia. Extremities were normal. Nails of all fingers and toes were dystrophic. Nipples were normal. He had one single kidney. There was no history of malformation or skin abnormalities in the family.
Patient 2, a 25-year-old female, was the third child of healthy unrelated parents. Absent labia minor, tight vagina and small uterus were noted at birth. She had surgery for obstruction of the vagina and lacrymal ducts in the first year of life, and for bilateral cataract at 20 years of age. She had an unexplained rise in temperature during childhood. She presented blepharophymosis, a narrow and pinched nose, a small mouth with a thin superior lip and normal uvula, and there was evidence of hypohydrotic ectodermal dysplasia: skin of the face was thin and dry, scalp hair was easily breakable, nails were dysplastic, hair at the lateral side of the eyebrows, eyelids, axilla and pubic region were sparse and there was hypodontia (19 teeth) with enamel and dentine dysplasia, and persistence of four deciduous teeth. Breast development was normal. Examination of the extremities revealed a minor loss of substance on the cubital side of the third phalanx of the left fifth finger, a minimal medial cleft of the third phalanx of the right fifth finger, and a cutaneous complete three to four syndactyly of the right feet. There was no history of malformation or skin abnormalities in the family. The clinical findings were most consistent with the diagnosis of the RHS type of ectodermal dysplasia (Table 1).
The 16 exons of the TP63 gene were PCR-amplified from genomic DNA, using primers described elsewhere,16 and directly sequenced using the PRISM Ampli Taq FS Ready Reaction Dye Terminators sequencing kit (PE Applied Biosystems) and a PE Applied Biosystems 377 automated DNA sequencer, as previously described.14
The R279H mutation was introduced by site-directed mutagenesis, using the QuickChange XL kit (Stratagene), into the different human TP63 cDNAs (TAp63α, TAp63γ, ΔNp63α, ΔNp63γ) cloned into the pCDNA3 vector (Invitrogen). For transactivation assays, 2.5 × 105 Saos-2 cells, plated in six-well plates, were transfected using the FuGENE 6 transfection reagent (Roche), with 1 μg of PG13-Luc reporter plasmid, different amounts of TP63 expression vectors and 2 μg of the pC53-SN plasmid, expressing the wild-type form of human TP53. A constitutive luciferase expression vector (pRL-CMV, 100 ng) was used in all samples to normalize for transfection efficiency and sample preparation. Cells were lysed 48 h after the transfection and assayed for transactivation using the Dual-luciferase Reporter Assay System (Promega). All experiments were performed in duplicate.
Results
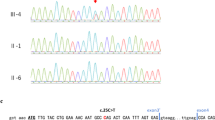
In patient 1, we detected at codon 570, within exon 14, a 1 bp deletion (1709DelA), resulting in a stop codon 22 amino-acids downstream the normal stop codon (Figure 2a). In patient 2, TP63 sequence analysis revealed, within exon 7, at codon 279 (numbered according to the TAp63α isoform, Genbank accession AF075430) a CGC to CAC transition resulting into the substitution of a conserved arginine by an histidine (Figure 2b).

Detection of heterozygous TP63 mutations in patients with RHS. The TP63 gene was PCR-amplified from genomic DNA and directly sequenced. Mutation or deletion are indicated by an arrow. (a) 1709DelA frameshift deletion (exon 14) in patient 1. (b) Heterozygous R279H mutation (exon 7) in patient 2. The predicted amino-acid change is indicated above the nucleotide sequence. In each panel, sequences observed in the proband (bottom) and in a control (top) are shown.
Functional analysis of the R279H mutation, performed in vitro, showed that this missense mutation did not decrease the transcriptional activity of the TAp63γ isoform on a TP53 reporter system (Figure 3a) but disrupted the dominant negative activity of the ΔN α and γ TP63 isoforms on the transcriptional activity of TP53 (Figure 3b).

Functional characterization of the TP63 R279H mutation using a TP53 reporter system. (a) Analysis of the transcriptional activity of wild-type and mutant TP63 isoforms. Human Saos-2 cells were cotransfected with 1 μg of PG13-Luc reporter plasmid and 2 μg of wild-type TP53 expression vector (black) or with 2 μg of wild-type (wt; white) or mutant (R279H; gray) TAp63α or γ expression vectors. The relative transactivation activity is given with respect to TP53 expression vector. (b) Analysis of the repressing activity of the wild-type and mutant ΔN isoforms. Human Saos-2 cells were cotransfected with 1 μg of PG13-Luc reporter plasmid and 2 μg of TP53 expression vector together with 2 (1:1 ratio with respect to TP53), 4 (1:2) or 10 (1:5) μg of wild-type (wt; white) or mutant (R279H; gray) ΔNp63α or γ expression vectors. pCDNA3 vector was used to normalize for DNA amount to 13.1 μg in each sample. The relative transactivation activity is given with respect to ΔNp63α expression vector. Error bars indicate SD in duplicate experiments.
Discussion
Here we report, in two patients with RHS, mutations within the TP63 gene, suggesting that RHS is part of the spectrum of phenotypes associated with mutations within this gene. Yet, we believe that RHS was the most likely diagnosis in both patients since they did not present either ankyloblepharon or ectrodactyly, which argues against the diagnosis of AEC and EEC, respectively (Table 1). Absence of cleft palate and presence of syndactyly and of genital malformations observed in patient 2 had already been reported in families with RHS.3,5
In the two patients, we identified one missense and one frameshift TP63 mutations, respectively. The exact function of the TP63 protein has not been established so far and the numerous TP63 isoforms make it difficult to predict the biological consequences of the mutations. Indeed, the TP63 gene, composed of 16 exons (Figure 1a), encodes six main isoforms (Figure 1b). The usage of an alternative promoter results in isoforms containing or lacking a transactivation domain (TA) at their NH2-terminus and respectively designed TAp63 and ΔNp63 (for a review see van Bohkoven and Brunner18). An alternative splicing produces three different C-termini, α, β, γ. All isoforms contain a DNA binding domain and a tetramerization domain sharing, respectively 65 and 35% of homology with that of the TP53 protein and the α isoforms contain an additional tail with a sterile alpha motif (SAM), involved in protein–protein interactions, and a post-SAM transactivation inhibitory domain (TID). The TA isoforms are able to activate in vitro TP53 target genes and the ΔN isoforms have dominant activities and constitute transcriptional repressors.17 The mutation identified in patient 1, 1709DelA, located in the post-SAM region is predicted to affect only the TP63α isoforms. The R279H mutation, that we identified in patient 2, appears to be a recurrent TP63 mutation that had been reported in several EEC unrelated patients.11,13,15,18,19 Interestingly, this residue corresponds to the R248 hot-spot mutation in TP53. Like all (except one) TP63 mutations reported in families with the EEC syndrome, this mutation occurs within the DNA-binding domain present within all the TP63 isoforms. We found that the R279H mutation inactivated the repressing activity of the ΔN α and γ isoforms (Figure 3b), but surprisingly had no effect on the transcriptional activity of TAp63γ (Figure 3a). We had previously observed that the TP63 R204Q mutation, detected in a patient with EEC, did not alter the transcriptional activity of TAp63γ (unpublished results) and this had already been reported for the ADULT R298Q mutation.20 These data suggest that certain TP63 mutant proteins retain their ability to bind DNA. It is interesting to note that these two mutations detected in RHS, like most of the other TP63 mutations described within the EEC and EEC-like syndromes, have in common to alter the transdominant negative effect of ΔNp63α isoform toward TP53. Therefore, our data suggest that the transdominant negative effect of ΔNp63 toward TP53 does not result from a competition in the DNA binding.
The identification of missense TP63 mutations within the DNA binding domain in EEC and of missense mutations within the SAM domain in AEC had suggested a genotype–phenotype correlation. In contrast, mutations in the SAM and post-SAM domains, particularly mutations impairing the open reading frame, appear to cause a variety of phenotypes including EEC, LMS, AEC, SHFM and, as shown by this report, RHS.
In conclusion, this report demonstrates, on a molecular basis, that RHS is also an EEC-like syndrome and thus represents the sixth EEC-like developmental disorder resulting from alteration of the TP63 gene, which highlights the wide phenotypic spectrum of TP63 mutations.
References
Rapp RS, Hodgkin WE : Anhydrotic ectodermal dysplasia: autosomal dominant inheritance with palate and lip anomalies. J Med Genet 1968; 5: 269–272.
Silengo MC, Davi GF, Bianco R et al: Distinctive hair changes (pili torti) in Rapp–Hodgkin ectodermal dysplasia syndrome. Clin Genet 1982; 21: 297–300.
Schroeder Jr HW, Sybert VP : Rapp–Hodgkin ectodermal dysplasia. J Pediat 1987; 110: 72–75.
Salinas CF, Montes-G GM : Rapp–Hodgkin syndrome: observations on ten cases and characteristic hair changes (pili canaliculi). Birth Defects Orig Art Ser 1988; 24: 149–168.
Rodini ES, Freitas JA, Richieri-Costa A : Rapp–Hodgkin syndrome: report of a Brazilian family. Am J Med Genet 1990; 36: 463–466.
Breslau-Siderius EJ, Lavrijsen APM, Otten FWA, van der Schroeff JG, Swart JGN : The Rapp–Hodgkin syndrome. Am J Med Genet 1991; 38: 107–110.
Walpole IR, Goldblatt J : Rapp–Hodgkin hypohydrotic ectodermal dysplasia syndrome. Clin Genet 1991; 39: 114–120.
Hart TC, Kyrkanides S : Cephalometric analysis of Rapp–Hodgkin syndrome. J Med Genet 1994; 31: 758–760.
Moerman P, Fryns JP : Ectodermal dysplasia, Rapp–Hodgkin type in a mother and severe ectrodactyly-ectodermal dysplasia-clefting syndrome (EEC) in her child. Am J Med Genet 1996; 63: 479–481.
Cambiaghi S, Tadini G, Barbareschi M, Menni S, Caputo R : Rapp–Hodgkin syndrome and AEC syndrome: are they the same entity? Br J Dermatol 1994; 130: 97–101.
Celli J, Duijf P, Hamel BC et al: Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 1999; 99: 143–153.
McGrath JA, Duijf PHG, Doetsch V et al: Hay–Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet 2001; 10: 221–229.
Van Bohkoven H, Hamel BJC, Bamshad M et al: p63 gene mutations in EEC syndrome, limb–mammary syndrome, and isolated split hand–split foot malformation suggest a genotype–phenotype correlation. Am J Hum Genet 2001; 69: 481–492.
Amiel J, Bougeard G, Francannet C et al: TP63 gene mutation in ADULT syndrome. Eur J Hum Genet 2001; 9: 642–645.
Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P : Split-hand/split–foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet 2000; 67: 59–66.
Hagiwara K, McMenamin MG, Miura K, Harris CC : Mutational analysis of the p63/p73L/p51/p40/CUSP/KET gene in human cancer cell lines using intronic primers. Cancer Res 1999; 59: 4165–4169.
Yang A, Kaghad M, Wang Y et al: p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998; 2: 305–316.
Van Bohkoven H, Brunner HG : Splitting p63. Am J Hum Genet 2002; 71: 1–13.
South AP, Ashton GH, Willoughby C et al: EEC (ectrodactyly, ectodermal dysplasia, clefting) syndrome: heterozygous mutation in the p63 gene (R279H) and DNA-based prenatal diagnosis. Br J Dermatol 2002; 146: 216–220.
Duijf PHG, Vanmolkot KRJ, Propping P et al: Gain-of-function mutation in ADULT syndrome reveals the presence of a second transactivation domain in p63. Hum Mol Genet 2002; 11: 799–804.
Acknowledgements
We are grateful to Françoise Charbonnier for technical support, to Bert Vogelstein who provided us the PG13-Luciferase plasmid and to Chikashi Ishioka who provided us the pCI51 yeast plasmid containing the human TAp63γ cDNA. We are indebted to Mario Tosi for critical review of the manuscript. This work was supported by l'Association pour la Recherche sur le Cancer, la Fondation pour la Recherche Médicale and la Ligue Nationale contre le Cancer. Gaëlle Bougeard was supported by a grant from le Ministère de la Recherche and from l'Association pour la Recherche sur le Cancer.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bougeard, G., Hadj-Rabia, S., Faivre, L. et al. The Rapp–Hodgkin syndrome results from mutations of the TP63 gene. Eur J Hum Genet 11, 700–704 (2003). https://doi.org/10.1038/sj.ejhg.5201004
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201004
Keywords
This article is cited by
-
Novel HOXD13 variants in syndactyly type 1b and type 1c, and a new spectrum of TP63-related disorders
Journal of Human Genetics (2022)
-
Taurodontism in dental genetics
BDJ Open (2021)
-
A new mutation in TP63 is associated with age-related pathology
European Journal of Human Genetics (2007)
-
Klinische Variabilität bei Mutationen im p63-Gen
Monatsschrift Kinderheilkunde (2005)
