
Abstract
We evaluated the effects of polymorphic markers within the β globin gene cluster on HbF expression in two groups. These groups were randomly selected from a survey of HbF distribution in a large population study of unrelated healthy Algerian adults (n=827). The first group contained individuals with normal HbF levels (0.1–0.5%) and the second group contained individuals with raised HbF levels (0.8–2.3%). Of the various polymorphic markers analysed, only the −309 Gγ A→G, the −158 Gγ C→T, the Gγ IVS2 TC (TG)9 AG (TG)2 (CG)2 and the −540 β (AT)9 T5 sequence configurations were significantly associated with increased HbF levels. More than 84% of the subjects with elevated HbF levels carried one or several of these four marker configurations, suggesting that the β globin gene cluster exerts a significant effect on HbF expression in healthy individuals.
Similar content being viewed by others

Introduction
The developmental switch from foetal (α2 γ2) to adult (α2 β2) haemoglobin (Hb) occurs just before birth. In normal subjects: (i) foetal Hb (HbF) constitutes less than 1% of the total Hb by the end of the first year of life; (ii) the synthesis of HbF is restricted to a subpopulation of red cells, known as F-cells (FC),1,2 (iii) HbF levels are directly correlated to the number of FC.3,4 Hereditary Persistence of Foetal Haemoglobin (HPFH) is characterised by persistently high levels of HbF after birth. Large deletions of varying sizes within the β globin cluster and single base substitutions in the promoter region of one of the two γ globin genes result in HPFH, with HbF in the range of 5–30% in heterozygous individuals.5 These rare forms of HPFH show a clear Mendelian pattern of inheritance and HbF is uniformly distributed among all red blood cells (pancellular HPFH). In the more common form of HPFH, known as Swiss-type HPFH6 or heterocellular HPFH, HbF levels only increase slightly (1–4%). This form is genetically heterogeneous and seem to result from complex interactions between a variety of genetic determinants, some of which are linked to the β globin gene cluster. Studies have mapped some of the determinants to chromosomes Xp22.27 and 6q23,8,9 but the precise loci involved have not yet been identified.
The degree to which the β globin gene cluster contributes to HbF expression is still under debate.10,11 Some studies have suggested that this cluster is involved in the variability of HbF expression and FC number in individuals with one (heterozygote) or two copies of the β thalassaemic (β thal) or the sickle cell gene, whereas others failed to confirm these findings.12,13 Studies of sequence variations in the Locus Control Region hypersensitive site-2 (β LCR 5′HS2),14,15,16 the second intron of the A γ gene (Aγ IVS2),17 the promoter region of the β globin gene18,19 and the -158 5′ of the Gγ gene20,21 have found a genetic association with HbF levels, FC number or both. These association studies have so far been performed exclusively on populations subjected to different degrees of anaemic stress or on carriers of various β globin gene defects.
The aim of this study was to determine the effect of these putative genetic markers on HbF expression in healthy adults free from any globin gene defects. We studied seven polymorphic regions of the β globin gene cluster, including four repeat sequences, and evaluated their association with HbF expression in healthy Algerian adults. Our data suggest strongly that there is a significant association between markers of the β globin gene cluster and HbF levels in healthy adults.
Materials and methods
Haematology and haemoglobin analysis
Haematological parameters were measured by use of automated counters (STKB Coultronic, Margency, France). HbA2 and HbF levels were estimated by use of a highly sensitive HPLC method.22
Population survey for HbF distribution
Informed consent was obtained from 859 unrelated Algerian adults. Their age ranged from 18 to 60 years and around 90% of them were younger than 40 years. Thirty-two of the subjects were carriers of S, C or the β thalassemia trait and were excluded from the study. The HbF level was measured in the remaining 827 healthy adults (417 males and 410-non pregnant females).
DNA analysis
Genomic DNA was extracted from the peripheral blood leukocytes of 97 randomly selected subjects: 51 with raised HbF levels and 46 with normal HbF levels. We analysed the entire β globin gene segment and the foetal globin gene promoter region for mutations by use of denaturing gradient gel electrophoresis (DGGE).23,24 Deletions in the α globin gene locus were sought by a PCR-based procedure.25
The following polymorphisms were investigated by use of previously described PCR-based procedures and, when necessary, by sequencing the cloned PCR products. The (AT)x N12 (AT)y repeat configurations within the 5′HS2 of β LCR,14 the (TG)n (CG)m configurations in the IVS2 Gγ and Aγ,17 the (AT)x Ty repeat configuration in the 5′ β globin gene, were characterised by size separation and, when appropriate, by direct nucleotide sequencing.26 The sequence variations, C→G at -369; A→G at -309 and C→T at -158 in the Gγ promoter region were explored by PCR-RFLP using BfaI, MboII and XmnI enzymes, respectively.
Statistical analysis
Due to the large size of the study population (males=417; females=410), the Z test27 was used to compare the mean HbF levels of the two genders. The 2×2 χ test was performed to detect differences in the distribution of repeat sequence configurations and single nucleotide polymorphisms between the phenotype groups. Statistical significance was set at P<0.05.
Given that some of our genotypes have an unknown gametic phase and include a large number of alleles, Arlequin, a program for the analysis of population genetic data, was used (http://lgb.unige.ch/arlequin) to perform a likelihood method for the analysis of linkage disequilibrium between the pairs of genetic marker configurations in each subgroup and also to establish the maximum likelihood haplotype frequency using an Expectation-Maximization (E.M) algorithm.28 The relationship between absolute HbF levels and genetic markers was investigated by use of the Students' t-test.
Results
HbF levels in the population
We studied 827 healthy adults (417 males and 410 females) with normal haematologic and Hb profiles. HbF levels ranged between 0.1 and 2.3%. In 520 subjects (62.9%), HbF levels were between 0.2 and 0.3%. In 86 subjects (10.4%) HbF levels were equal or above 0.8%. In 47 of these subjects (54.7%), HbF levels were ⩾1%. The average HbF level in the population was 0.39±0.29% (mean±SD). A significant difference (P<0.01; Z test) was noted between males (HbF, 0.35±0.26%) and females (HbF, 0.435±0.312%). A higher proportion of females had increased HbF levels.
Phenotype analysis
We randomly selected 97 subjects for genetic studies: 51 with raised HbF levels (0.8–2.3%; High F group) and 46 with normal range of HbF levels (0.1–0.5%; NRF group). The haematologic indices of these 97 subjects were in the normal range. The means±SD of the High F and NRF subjects were respectively 2.58±0.26% and 2.54±0.25% for HbA2; 14 g/dl±1.12 and 13.42 g/dl±1.43 for total Hb; 41.56%±3.34 and 39.27±4.30% for hematocrit; 85.8 fl±5.06 and 86.15 fl±5.49 for MCV, and 29g/dl±1.88 and 29.98g/dl±2.55 for MCH. These haematologic indices suggest that the β thalassaemic allele is absent in these subjects. This was further confirmed by DNA analysis using a variety of techniques, including nucleotide sequencing when appropriate.
Marker analysis
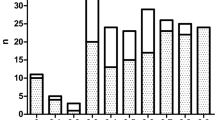
The DNA regions analysed and the sequence configurations encountered in this study are listed in Figure 1. The (AT)x N12 (AT)y configurations of β LCR HS2 were named A to J, the (TG)n (CG)m configurations of Gγ IVS2 were named K to S, the (TG)n (CG)m configurations of Aγ IVS2 were named T and U and the (AT)x Ty configurations 5′ of β were named V to Z. The allele and genotype frequencies for each site are given in Tables 1a and b , respectively. The frequency of the repeat sequence configurations and simple nucleotide variations whose distributions differed significantly between the High F and NRF groups are given in Figure 2.

βLCR 5′ HS2, Gγ, Aγ IVS2 and 5′ of β globin gene sequence configurations in NRF and High F subjects. The configurations of LCR 5′HS2 were named A to J, those of Gγ IVS2 were named K to S, those of Aγ IVS2 were named T and U and those 5′ of β globin were named V to Z. A, K, T and V are the reference sequence configurations from the HUMHBB reference sequence. The nucleotides are numbered according to the HUMHBB sequence.

Distribution of the polymorphic markers among High F and NRF groups. Filled bars: high F group; empty bars: normal range of the HbF group (NRF), *: statistically significant, P: P value.
Gγ and Aγ promoter regions
None of the individuals in the High F group harboured any sequence changes in the Gγ and Aγ promoter regions (up to −264 bp), except for the common C→T polymorphism at −158 of Gγ and the 4 bp deletion upstream of Aγ (−222 to −225). The G at −369 5′ of Gγ was not polymorphic in either of the groups, whereas the A→G substitution at −309 was only found in the High F group (n=9). The frequency of the −158 Gγ C→T polymorphism (detected as XmnI- →XmnI+polymorphism) was statistically different between the two groups (P= 0.02): XmnI+ was present in 51% of High F subjects vs 28% in the NRF group (Figure 2).
Configurations of the (AT)x N12 (AT)y motif in β LCR HS2
Ten different sequence configurations of the (AT)x N12 (AT)y motif were found among the 194 chromosomes studied. The most common configuration in both groups was the A configuration (Table 1a), which is the reference sequence (GenBank coordinates: 8881-8934). Although the configuration D was much more common in the High F group (52%) than in the NRF group (32.6%) (Table 1b), the difference was not statistically significant (P=0.06).
Gγ IVS2 (TG)n (CG)m configurations
In the Gγ IVS2, two novel sequence configurations for (TG)n (CG)m motifs (configurations Q and R) were found in three High F subjects. The prevalence of configurations M and O differed significantly between the two groups: 25.5% of the High F subjects carried configuration M vs 2.27% in the NRF group (P=0.002) and 40.9% of the NRF subjects carried configuration O compared to 5.76% in the High F group (P=0.0001) (Figure 2).
5' β globin gene promoter (AT)x (T)y motif
Configuration V (reference sequence) was the most frequent configuration in both two groups (Table 1a), but the configuration W was significantly more common in the High F group than in the NRF group (P=0.012) (Figure 2).
Marker combinations
Over 84% of the High F subjects (43/51) harboured at least one of four sequence configurations, namely G at −309 of Gγ, XmnI+, M and W configurations, compared to only 43% (20/46) of the NRF group (Table 2). We found that 22 High F and 17 NRF subjects carried one of these four sequence configurations. Significantly, more of the subjects in the High F group (37.5 vs 6.5%) contained two configurations. None of the NRF group subjects had three configurations.
Arlequin tests
Table 3 summarises the results of the linkage disequilibrium test between pairs of genetic markers in High F subjects. This test showed linkage disequilibrium (i) between configurations D (β LCR-HS2) and M (Gγ IVS2) and between the XmnI+ polymorphism and configurations D, M and W (−540 of β globin gene). However, configuration W was not associated with either D or M. The Arlequin data confirmed that the XmnI+ site is in strict linkage disequilibrium with configuration T in Aγ IVS2 as demonstrated earlier,18 which validates our statistical testing procedure. The maximum likelihood haplotype frequency test failed to reveal any specific associations between the β globin gene cluster haplotypes (constituted by all the studied markers) and the HbF phenotype groups.
Genotype/phenotype association
Since more than 84% of the High F subjects had at least one of the four sequence configurations compared to 43% of the NRF subjects, we further investigated their combined association with HbF expression for all the studied individuals (Table 4). Our data revealed that subjects carrying one, two or three configurations had significantly higher mean absolute HbF values (g/dl) than those not carrying any of these (P<0.001). Furthermore, the effect on HbF level was more pronounced when two markers were present than when only one was present (P<0.005).
Discussion
The amount of HbF in normal adults varied by more than 20-fold. The distribution curve was skewed towards right with around 10% of the subjects having HbF levels above 0.8%. Family studies have shown that a strong genetic component determines the HbF level in subjects at the extremes of this spectrum.3,4 This is the reason why we chose subjects at the extreme ends of the spectrum for the further genetic analysis (NRF and High F groups).
None of these subjects harboured any β globin gene defects or any anomalies in the foetal globin gene regions. We studied seven polymorphic markers spread through the 40 Kb segment of β globin gene cluster and assessed their association with HbF expression. Four polymorphisms (−309 Gγ A→G, −158 Gγ C→T (XmnI+), TC (TG)9 AG (TG)2 (CG)2 in Gγ IVS2 and (AT)9 T5 5′ of the β globin gene) showed a statistically significant independent association with the HbF expression. The −309 Gγ A→G polymorphism was found exclusively in High F subjects. These specific sequence configurations were found in more than 84% of the high F subjects and only in 43% of the subjects with HbF in the normal range.
It is interesting that the −309 Gγ A→G variation was only found in the High F group because we (unpublished observations) and others16,29 have shown that this variation is specific to the Benin βS haplotype, which is usually associated with low HbF expression. In our study, none of the subjects with −309 Gγ A→G variation had this haplotype and no other population data is available so we can not determine whether, in a context different from the Benin βS haplotype, this polymorphism plays a role in HbF expression. Similarly, the Gγ IVS2 TC (TG)9 AG (TG)2 (CG)2 configuration was previously found on two chromosomes associated with the Benin βS haplotype.29 Our findings show that these two polymorphic markers are also present in normal chromosomes and are associated with raised HbF levels. Conversely, the XmnI+ is a common polymorphism that has been extensively explored in different populations and has been shown to influence the number of FC and HbF levels in normal30,31 and anaemic subjects.20,21
The (AT)9 T5 configuration (configuration W), which is located within a putative transcriptional silencer32 of the β globin gene, was originally described in a silent β thalassaemia case33 and has been shown to be associated with reduced sickle gene expression.34 However, another study involving a few non-anaemic subjects, did not confirm this effect on β globin gene expression.12 Our data, based on a much larger number of subjects, reveal that this configuration is associated with raised HbF levels. Genes that have minor effects cannot be studied on an individual basis, but their effects may be revealed in a statistical manner through a population study. This might explain why some studies with small number of individuals failed to demonstrate this association.
Of the 22 High F subjects with the (AT)9 T5 sequence configuration, 15 had XmnI+. The co-presence of these sequences both in cis and in trans is associated with elevated HbF levels in homozygous and heterozygous β thalassaemia.18,19 Our data suggest that even in the absence of any β globin defects, XmnI+ and (AT)9 T5, are strongly associated with elevated levels of HbF.
As more than 84% of individuals (43/51) carry at least one of the four marker configurations and as those possessing one of these marker configurations have higher mean HbF levels than those lacking them, these markers may contribute to HbF expression in an independent manner. We cannot totally exclude the possibility that these markers have a direct effect, as they are located in evolutionarily conserved regulatory regions of the globin genes. Alternatively they may act indirectly, as a marker in linkage disequilibrium with an unidentified sequence in the region surrounding this cluster.
This is the first elaborate analysis to have explored the contribution of the β globin gene cluster in the expression of HbF in healthy adults, free of any globin gene defects. We have demonstrated that this cluster makes a significant genetic contribution to HbF.
References
Boyer SH, Belding TK, Margolet L, Noyes AN . Fetal hemoglobin restriction to a few erythrocytes (F cells) in normal human adults Science 1975 188: 361–363
Wood WG, Stamatoyannopoulos G, Lim G, Nute PE . F cells in the adult: normal values and levels in individuals with hereditary and acquired elevation of HbF Blood 1975 46: 671–687
Zago MA, Wood WG, Clegg JB, Weatherall DJ, O'Sullivan M, Gunson H . Genetic control of F cells in human adults Blood 1979 53: 977–986
Miyoshi K, Kaneto Y, Kawai H et al. X-linked dominant control of F-cells in normal adult life: characterization of the Swiss type as hereditary persistence of foetal hemoglobin regulated dominantly by gene(s) on X chromosome Blood 1988 72: 1854–1860
Wood WG . Hereditary persistence of fetal hemoglobin and δβ thalassemia in Steinberg MH, Forget BG, Higgs DR and Nagel RL (eds) Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management 2001 Cambridge University Press pp 356–388
Marti HR . Normale und anormale mensliche Haemoglobine Berlin, Germany, Springer Verlag 1963
Dover GJ, Smith KD, Chang YC et al. Foetal hemoglobin levels in sickle cell disease and normal individuals are partially controlled by an X-linked gene located at Xp22.2 Blood 1992 80: 816–824
Craig JE, Rochette J, Fisher CA et al. Dissecting the loci controlling foetal haemoglobin production on chromosomes 11p and 6q by the regressive approach Nat Genet 1996 12: 58–64
Garner C, Mitchell J, Hatzis T, Reittie J, Farrall M, Thein SL . Haplotype mapping of a major quantitative-trait locus for fetal hemoglobin production, on chromosome 6q23 Am J Hum Genet 1998 62: 1468–1474
Efremov GD, Gjorgovski I, Stojanovski N et al. One haplotype is associated with the Swiss type of hereditary persistence of fetal hemoglobin in the Yugoslavian population Hum Genet 1987 77: 132–136
Leonova J, Kazanetz EG, Smetanina NS, Adekile AD, Efremov GD, Huisman TH . Variability in the fetal hemoglobin level of the normal adult Am J Hematol 1996 53: 59–65
Galanello R, Meloni A, Gasperini D et al. The repeated sequence (AT)x (T)y upstream to the beta-globin gene is a simple polymorphism Blood 1993 81: 1974–1975
Lu ZH, Steinberg MH . Fetal hemoglobin in sickle cell anemia: relation to regulatory sequences cis to the beta-globin gene. Multicenter Study of Hydroxyurea Blood 1996 87: 1604–1611
Perichon B, Ragusa A, Lapoumeroulie C et al. Inter-ethnic polymorphism of the beta-globin gene locus control region (LCR) in sickle-cell anemia patients Hum Genet 1993 91: 464–468
Merghoub T, Maier-Redelsperger M, Labie D et al. Variation of fetal hemoglobin and F-cell number with the LCR-HS2 polymorphism in nonanemic individuals Blood 1996 87: 2607–2609
Oner C, Dimovski AJ, Altay C et al. Sequence variations in the 5′ hypersensitive site-2 of the locus control region of beta S chromosomes are associated with different levels of fetal globin in hemoglobin S homozygotes Blood 1992 79: 813–819
Lapoumeroulie C, Castiglia L, Ruberto C et al. Genetic variations in human fetal globin gene microsatellites and their functional relevance Hum Genet 1999 104: 307–314
Ragusa A, Lombardo M, Beldjord C et al. Genetic epidemiology of beta-thalassemia in Sicily: do sequences 5′ to the G gamma gene and 5′ to the beta gene interact to enhance HbF expression in beta-thalassemia? Am J Hematol 1992 40: 199–206
Goncalves I, Ducrocq R, Lavinha J et al. Combined effect of two different polymorphic sequences within the beta globin gene cluster on the level of HbF Am J Hematol 1998 57: 269–276
Gilman JG, Huisman TH . DNA sequence variation associated with elevated fetal G gamma globin production Blood 1985 66: 783–787
Labie D, Pagnier J, Lapoumeroulie C et al. Common haplotype dependency of high G gamma-globin gene expression and high Hb F levels in beta-thalassemia and sickle cell anemia patients Proc Natl Acad Sci U S A 1985 82: 2111–2114
Bisse E, Wieland H . High-performance liquid chromatographic separation of human haemoglobins. Simultaneous quantitation of foetal and glycated haemoglobins J Chromatogr 1988 434: 95–110
Gottardi E, Losekoot M, Fodde R, Saglio G, Camaschella C, Bernini LF . Rapid identification by denaturing gradient gel electrophoresis of mutations in the gamma-globin gene promoters in non-deletion type HPFH Br J Haematol 1992 80: 533–538
Ghanem N, Girodon E, Vidaud M et al. A comprehensive scanning method for rapid detection of beta-globin gene mutations and polymorphisms Hum Mutat 1992 1: 229–239
Baysal E, Huisman TH . Detection of common deletional alpha-thalassemia determinants by PCR Am J Hematol 1994 46: 208–213
Trabuchet G, Elion J, Baudot G et al. Origin and spread of beta-globin gene mutations in India, Africa, and Mediterranea: analysis of the 5′ flanking and intragenic sequences of beta S and beta C genes Hum Biol 1991 63: 241–252
Scherrer B . Biostatistique G Morin (ed) 1984 Quebec
Excoffier L, Slatkin M . Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population Mol Biol Evol 1995 12: 921–927
Lanclos KD, Oner C, Dimovski AJ, Gu YC, Huisman TH . Sequence variations in the 5′ flanking and IVS-II regions of the G gamma- and A gamma-globin genes of beta S chromosomes with five different haplotypes Blood 1991 77: 2488–2496
Sampietro M, Thein SL, Contreras M, Pazmany L . Variation of HbF and F-cell number with the G-gamma XmnI (C→T) polymorphism in normal individuals Blood 1992 79: 832–833
Garner C, Tatu T, Reittie JE et al. Genetic influences on F cells and other hematologic variables: a twin heritability study Blood 2000 95: 342–346
Berg P, Williams D, Qian R et al. A common protein binds to two silencers 5′ to the human beta-globin gene Nucleic Acids Res 1989 17: 8833–8852
Semenza GL, Delgrosso K, Poncz M, Malladi P, Schwartz E, Surrey S . The silent carrier allele: beta thalassemia without a mutation in the beta-globin gene or its immediate flanking regions Cell 1984 39: 123–128
Zeng FY, Rodgers GP, Huang SZ et al. Sequence of the −530 region of the beta-globin gene of sickle cell anemia patients with the Arabian haplotype Hum Mutat 1994 3: 163–165
Acknowledgements
This work was supported by grant N˚ TS3*-CT93-0244DG12HSMU from the European Union. S Zertal-Zidani is recipient of a fellowship from the ‘Association française de recherche génétique’.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zertal-Zidani, S., Ducrocq, R., Sahbatou, M. et al. Foetal haemoglobin in normal healthy adults: relationship with polymorphic sequences cis to the β globin gene. Eur J Hum Genet 10, 320–326 (2002). https://doi.org/10.1038/sj.ejhg.5200809
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200809
Keywords
This article is cited by
-
Contribution of β-globin cluster polymorphisms to raise fetal hemoglobin levels in normal adults
Molecular Biology Reports (2012)
