
Abstract
Tissue inhibitor of metalloproteinase-3 (TIMP-3) is a natural inhibitor of metalloproteinases involved in matrix degradation and ectodomain shedding of many cell-surface proteins, including death receptors and/or their ligands. In the present study, we examined the role of TIMP-3 in Fas-mediated neuronal cell death following cerebral ischemia, using both gene deletion and pharmacological approaches. In culture, exposure of primary cortical neurons to 2 h of oxygen–glucose deprivation (OGD) resulted in delayed neuronal cell death that was dependent on activation of the death receptor, Fas. Cortical cultures derived from timp-3−/− mice displayed partial resistance against OGD-induced neuronal cell death and also displayed increased shedding of Fas ligand (FasL) into the culture media, compared to wild-type control cultures. Both the increased neuroprotection and increased FasL shedding in timp-3−/− cultures were reversed by addition of exogenous metalloproteinase inhibitors, recombinant TIMP-3 or GM6001. In vivo, timp-3−/− mice showed marked resistance to a brief (30 min) middle cerebral artery occlusion (MCAO), but were not protected against more severe lesions induced by 90 min of MCAO. These studies demonstrate that TIMP-3 facilitates Fas-mediated neuronal cell death following OGD and plays a pro-apoptotic role in mild cerebral ischemia.
Similar content being viewed by others


Main
Fas (APO-1/CD95) is a member of the tumor necrosis factor receptor (TNFR) superfamily of cell-surface death receptors that triggers caspase-dependent apoptosis in many cell types.1 Receptor activation following binding of the cognate ligand, Fas ligand (FasL), results in assembly of an intracellular death-inducing signaling complex (DISC), activation of caspase 8 and transduction of an apoptotic cascade. Although Fas signaling is historically recognized for its role in immune modulation, Fas and FasL are also expressed in the adult mammalian nervous system and are thought to contribute to cell death in CNS trauma,2 neurodegenerative disease,3 multiple sclerosis4 and ischemia.5, 6, 7, 8, 9 Following focal cerebral ischemia in rodents, Fas gene expression is upregulated as a downstream response to JunN-terminal kinase-3 (JNK-3) (stress-activated protein kinase) activation.10 Since inhibition of Fas signaling is neuroprotective in both in vivo and in vitro rodent models of cerebral ischemia,5, 11 Fas activation is considered a key trigger of delayed neuronal death following stroke.
Fas receptor signaling is modulated at several levels. For example, production of decoy receptors blunt extracellular ligand binding,12 and the production of intracellular proteins can interfere with caspase activation and apoptotic signaling cascades.13 In addition, proteolytic shedding of FasL ectodomain from the cell surface by metalloproteinase activity strongly influences cell sensitivity to Fas-mediated death.14, 15 In non-neuronal cells, proteolytic shedding of the FasL ectodomain from the cell surface leads to enhanced or diminished death-promoting activity depending on cell type.14, 16, 17 However, the role of proteolytic shedding of FasL in cerebral ischemia has not previously been investigated.
Tissue inhibitor of metalloproteinase-3 (TIMP-3) is a secreted protein with natural inhibitory activity against metalloproteinases involved in matrix degradation and shedding of many cell-surface proteins, including cell-surface death receptors and/or their ligands.18 TIMP-3 inhibits the activity of members of the matrix metalloproteinase (MMP), a disintegrin and metalloproteinase (ADAM), and ADAM with thrombospondin domain (ADAM-TS) families.18 Unlike the other TIMP family members (TIMP-1, -2, -4), TIMP-3 is tightly bound to proteoglycans, suggesting that TIMP-3 activity is confined mainly to the cell surface.19 TIMP-3 has been shown to play a role in the regulation of receptor-mediated cell death in various cell types through inhibition of metalloproteinase activity and stabilization of cell-surface death receptors.20, 21, 22, 23, 24, 25
Within the CNS, TIMP-3 is transiently expressed in embryonic cerebral cortex during the period of naturally occurring programmed cell death26 and is upregulated and colocalized with Fas in penumbral cortical neurons undergoing apoptosis following ischemia in the adult.9 In culture, TIMP-3 is constitutively expressed by embryonic cortical neurons25, 27 and is necessary for Fas-mediated apoptosis induced by the chemotherapeutic drug, doxorubicin.25 Here, we report that TIMP-3 also acts to facilitate Fas-mediated neuronal cell death following ischemia. We compared neuronal cell death in cortical cultures established from wild-type versus timp-3−/− mice following exposure to oxygen–glucose deprivation (OGD) in vitro and following mild focal cerebral ischemia in vivo. Our results demonstrate that TIMP-3 facilitates Fas-mediated neuronal apoptosis following OGD in culture and serves a proapoptotic function following mild focal cerebral ischemia in vivo.
Results
OGD generates nuclear condensation, caspase-3 activation and neuronal apoptosis in cultured cortical neurons
To investigate the convergent roles of TIMP-3 and death receptor signaling in neuronal injury following ischemia, we utilized a common in vitro model of OGD. Previous work has demonstrated that 2 h of OGD leads to delayed apoptotic neuronal death in murine cortical cultures, associated with activation of both caspases 8 and 3,28 and that neuronal apoptosis in response to OGD is dependent on death receptor activation.5
To characterize the time course of the OGD response, we exposed wild-type cortical cultures to 2 h of OGD or control conditions and assessed neuronal survival at 12, 24 or 48 h following return to normoxic conditions. OGD treatment resulted in 60% neuronal cell loss by 24 h, as assessed by NeuN immunoreactivity (Figure 1d), with no further cell loss at 48 h. Concomitant with the decline in NeuN-positive cells, OGD stimulated an increase in nuclear condensation, appearance of activated caspase 3 immunoreactivity (Figure 1a–c) and an approximate 60% increase in apoptosis, as determined by a cell death oligonucleosome enzyme-linked immunosorbent assay (ELISA) (Figure 1e). Consistent with apoptotic cell death, OGD-treated cultures did not release lactate dehydrogenase (LDH) (data not shown), indicating that caspase activation, nuclear condensation and histone-associated DNA fragments were released into the cytoplasm without the loss of plasma membrane integrity.

OGD generates nuclear condensation, caspase-3 activation and neuronal apoptosis in cultured wild-type cortical neurons. Primary cortical neuron cultures established from timp-3+/+ mice were exposed to either 2 h of control conditions or 2 h of OGD followed by return to normal culture conditions for 12, 24 or 48 h. Control cultures (left panels) or OGD cultures (right panels) labeled with NeuN antibody (a), DAPI nuclear dye (b) and activated caspase-3 immunofluorescence (c). (d) Time course of neuronal death following 2 h of OGD. (e) The degree of apoptosis generated by 2 h OGD followed by return to normal culture conditions for 24 h as assessed by Cell Death ELISA (n=6, from two separate experiments, *P<0.001, OGD versus control conditions)
OGD-induced neuronal apoptosis is dependent upon Fas activation
To determine whether the Fas death receptor pathway contributes to neuronal death under our experimental OGD conditions, we first asked whether Fas and/or FasL are expressed by cortical neurons in culture. Figure 2a depicts dual immunolabeling for Fas and the neuron-specific marker, microtubule-associated protein-2 (MAP-2). As illustrated in the merged images, cortical neurons uniformly express both Fas and FasL in culture. These data are consistent with previous reports demonstrating that Fas2, 6, 25, 27 and FasL2, 25, 27 are constitutively expressed by cultured rodent cortical neurons, and that expression of these death receptors and their ligands alone is not sufficient to induce neuronal death.

Fas and FasL are expressed by cortical neurons in culture, and OGD-induced neuronal apoptosis is dependent on Fas receptor activation. The cellular localization of Fas (a) and FasL (b) in unstimulated primary cortical cultures was assessed by immunofluorescence staining and colocalization with the neuronal marker, MAP-2. Primary cortical neuron cultures established from wild-type mice were incubated in the presence or absence of Fas-Fc (10 μg/ml) or anti-FasL (20 μg/ml) under normoxic control conditions (c) and during 2 h of OGD (d). Cultures were continuously exposed to both inhibitors for 24 h after return to normoxia, followed by immunofluorescence staining for NeuN and assessment of neuronal survival. The number of NeuN-positive cells was determined for each treatment group and data were normalized to control normoxic conditions (n=6 cultures per group, *P<0.001 compared to normoxic conditions, #P<0.01 compared to OGD plus IgG or OGD plus IgG-Fc)
To determine whether Fas activation is required for OGD-induced neuronal cell death, as previously demonstrated for doxorubicin-induced cell death,25 we incubated wild-type neuronal cultures with a function-blocking anti-FasL antibody or with an Fc fragment of human IgG1 fused to the ectodomain of Fas (Fas-Fc), during and after exposure to OGD. These inhibitors act as decoy receptors to block the interaction of endogenous FasL with its receptor. As shown in Figure 2c, neither inhibitor affected neuronal survival under control normoxic conditions. However, both anti-FasL and Fas-Fc provided marked neuroprotection against 2 h OGD, as assessed at 24 h, whereas incubation with control IgG or IgG-Fc had no significant effect (Figure 2d). These observations indicate that OGD-induced death is dependent upon Fas–FasL interaction in our experimental paradigm.
Cultured timp-3−/− cortical neurons display increased resistance to OGD
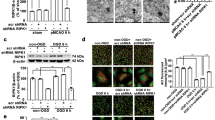
We previously demonstrated that rat cortical neurons constitutively express TIMP-3 mRNA and functional protein in culture,25 and have previously reported TIMP-3 mRNA expression by mouse cortical neurons in culture.27 Figure 3a demonstrates dual immunofluorescence labeling of wild-type cortical cultures for TIMP-3 and for the neuron-specific marker, NeuN. Merged images indicate that TIMP-3 is somatodendritically expressed. Western blot analyses on cortical culture cell lysates demonstrate TIMP-3 immunoreactive bands at approximately 50 and 21 kDa (Figure 3b). The calculated molecular weight of TIMP-3 is 21.6 kDa, and the 50-kDa band is likely to represent a TIMP-3 dimer. This antibody did not recognize recombinant TIMP (rTIMP)-1, 2 or 4 on western blots (data not shown).

Cultured timp-3−/− cortical neurons display increased resistance to OGD. The cellular localization of TIMP-3 in unstimulated wild-type primary cortical cultures was assessed by immunofluorescence staining and colocalization with the neuronal marker, NeuN (a), and by western blot analysis (b). (c) Primary cortical neuron cultures derived from timp-3+/+ (WT: filled bars) and timp-3−/− (KO: open bars) mice were exposed to 2 h of OGD followed by return to normal culture conditions for 24 h and assessment of neuronal survival by quantifying the number of NeuN-positive cells. Data are expressed as % neuronal survival normalized to control normoxic condition for each phenotype (n=6 from two separate experiments, *significantly different from timp-3+/+ WT cultures, P<0.001). (d) Cell death ELISA at 24 h post-OGD (n=6 from two separate experiments, *P<0.01 timp-3−/− versus timp-3+/+)
To test whether TIMP-3 is required for Fas-mediated neuronal cell death following OGD, we established cortical cultures from wild-type timp-3+/+ or knockout timp-3−/− embryos. Neuronal survival was similar in both genotypes under basal culture conditions as assessed at 8 days in vitro (124 608±5649 versus 102 468±4691 neurons in wild-type versus knockout cultures, respectively, mean±S.E.M., P=0.338), with 1–2% contamination with GFAP+ astrocytes (2.09±0.683 versus 1.8±0.62% GFAP+ cells in wild-type versus knockout cultures). Timp-3−/− cultures showed greater neuronal survival compared to the wild-type controls at both 24 and 48 h following 2 h exposure to OGD (Figure 3c). OGD treatment resulted in an approximate 53% neuron loss at 24 h in timp-3+/+ cultures, compared to 31% loss in timp-3−/− cultures. This was also confirmed by a cell death ELISA, indicating 20% less apoptosis in timp-3−/− cultures compared to wild-type controls at 24 h following OGD (Figure 3d).
rTIMP-3 and metalloproteinase inhibitors restore OGD sensitivity in timp-3−/− cultures
To ask whether the survival effects of TIMP-3 gene deletion are related to metalloproteinase disinhibition, we attempted to restore sensitivity of timp-3−/− neurons to OGD by phenotypic rescue with exogenous rTIMP-3 or with GM6001, a synthetic metalloproteinase inhibitor. As shown in Figure 4a, neither rTIMP-3 (1.25 μg/ml) nor GM6001 (25 μM) had any effect on neuronal survival in either strain under normoxic conditions, indicating that metalloproteinase inhibition per se is not neurotoxic. Neither rTIMP-3 nor GM6001 affected the sensitivity of timp-3+/+ cultures to OGD (Figure 4b). In contrast, both rTIMP-3 and GM6001 markedly increased OGD sensitivity in timp-3−/− cultures (Figure 4b). These data suggest that the proapoptotic activity of TIMP-3 under conditions of OGD is mediated through metalloproteinase inhibition and that timp-3−/− cultures show increased sensitivity to metalloproteinase inhibition under OGD conditions.

Restoration of OGD sensitivity in timp-3−/− cultures by metalloproteinase inhibitors. Primary cortical cultures established from timp-3+/+ and timp-3−/− mice were exposed to 2 h normoxia (a) or 2 h OGD (b) in the presence or absence of rTIMP-3 (1.25 μg/ml) or GM6001 (25 μM), followed by return to normal culture conditions for 24 h in the continued presence of the inhibitors. The number of NeuN-immunopositive cells was determined for each treatment group. Data are expressed as % neuronal survival as normalized to control normoxic conditions (n=6 from two separate experiments; *P<0.001, OGD timp-3+/+ versus normoxia timp-3+/+; #P<0.01 OGD timp-3−/− versus OGD timp-3+/+; $P<0.01 OGD timp-3−/− plus inhibitor versus OGD timp-3−/− without inhibitor)
Timp-3−/− neurons display enhanced proteolytic shedding of FasL in response to OGD
To determine whether the proapoptotic activity of TIMP-3 is related to stabilization of cell-surface FasL, we compared the levels of FasL released into the conditioned media by timp-3+/+ versus timp-3−/− cultures at 24 h after OGD. Under normoxic conditions, both timp-3+/+ and timp-3−/− cultures released very low to non-detectable levels of FasL into the conditioned media (0–4.3 pg/ml; Figure 5a). At 24 h following OGD, however, both timp-3+/+ and timp-3−/− cultures displayed increased shedding of FasL into the conditioned media, with timp-3−/− cultures releasing nearly three-fold more FasL into the media compared with that of timp-3+/+ cultures (21.07±9.52 versus 62.63±6.80 pg/ml, timp-3+/+ versus timp-3−/− conditioned medium, respectively; Figure 5b). The increased release of FasL into the culture medium in timp-3−/− cultures was blocked by exposure to the metalloproteinase inhibitor, GM6001, during and after OGD (Figure 5b), indicating that the increased FasL in the culture media was due to increased shedding rather than due to increased production of FasL. Thus, TIMP-3 gene deletion results in increased proteolytic activity at the cell surface in response to OGD, manifest by increased release of FasL into the culture media.

OGD-induced shedding of FasL and effects of sFasL and MMP-3 on neuronal survival. Cortical cultures established from timp-3+/+ or timp-3−/− mice were exposed to 2 h normoxia (a) or OGD (b) in the presence or absence of GM6001 (25 μM), followed by return to normoxic conditions in the continued presence of GM6001 for 24 h. FasL release into the conditioned media was assayed by FasL ELISA (n=4–6 per condition, *P<0.01 OGD versus normoxic conditions; #P<0.01, OGD timp-3−/− versus OGD timp-3+/+; $P<0.01 OGD timp-3−/− GM6001 versus OGD timp-3−/−). (c) Cortical cultures were incubated for 24 h in the presence or absence (control) of soluble FasL. (d) Cortical cultures were incubated in the presence or absence of active MMP-3 for 2 h during normoxia or OGD conditions and for 24 h thereafter (*P<0.01 compared to normoxia without MMP-3; #P<0.01 OGD MMP-3 versus OGD alone)
Previous studies have demonstrated that membrane-bound, but not soluble FasL, is neurotoxic for rat cortical neurons in culture.2, 25 Similarly, we found that soluble FasL was not toxic to murine cortical neurons following 24 h incubation with 5–50 ng/ml sFasL (Figure 5c). To determine whether increased proteolytic activity is neuroprotective against OGD, we incubated timp-3+/+ neuronal cultures with the active catalytic subunit of MMP-3 during and after the termination of OGD. MMP-3 alone was mildly neurotoxic under control normoxic conditions; however, incubation with active MMP-3 during and after OGD revealed a neuroprotective effect, such that MMP-3 increased neuronal survival by approximately 32% (Figure 5d). These results are consistent with previous studies demonstrating a neuroprotective action of MMP-3 against Fas-mediated neuronal cell death.25
Timp-3−/− mice display increased resistance to mild focal ischemia in vivo
Fas regulates cell death induced by focal ischemia in rodents5, 29 and is coexpressed with TIMP-3 in apoptotic neurons.9 To test whether TIMP-3 displays proapoptotic activity in vivo, we compared the sensitivity of wild-type and timp-3−/− mice to mild focal ischemia induced by transient occlusion of the middle cerebral artery (MCAO). For initial experiments, we utilized a model of mild cerebral ischemia in which delayed caspase-dependent apoptotic neuronal death predominates.30 Mice received 30 min of MCAO using the monofilament procedure, which resulted in a reduction of regional cerebral blood flow (rCBF) to approximately 20% of baseline in both strains (25±6.5 versus 23±1.9%, timp-3−/−versus wild type, respectively), with a return to approximately 60% of normal, at 30 min following filament removal as assessed using laser Doppler flowmetry (Figure 6b). Cerebral vasculature was visualized by India Ink perfusion and was similar in both timp-3−/− and wild-type mice (n=5 mice per strain) with patent Circle of Willis in both strains (Figure 6a). Physiological parameters were also similar between genotypes before, during and after MCAO and remained within normal limits in both strains (Table 1).

Comparison of cerebral vasculature and cerebral blood flow in timp-3+/+ and timp-3−/− mice. (a) Both timp-3+/+ and timp-3−/− have patent Circle of Willis. (b) Changes in cerebral blood flow in timp-3+/+ versus timp-3−/− mice during and after 30 min MCAO (n=5 mice per strain)
Ten mice from each genotype (timp-3+/+ or timp-3−/−) were subjected to 30 min of transient MCAO and killed after 3 days of reperfusion. Neurodegeneration was assessed using computer-assisted morphometric analysis of histological sections. Terminal deoxynucleotidyl transferase-mediated dUTP–biotin nick end-labeling (TUNEL)-positive cells corresponded with Fluoro-Jade staining in both strains (Figure 7a–d). Only five of the ten mice of each genotype met our selection criteria of displaying a lesion at 3 days, despite rCBF reductions to approximately 20% baseline in all animals. This suggests that both genotypes required equivalent ischemic thresholds to induce cell death following brief transient MCAO. In the mice that demonstrated infarct (n=5 per genotype), both average lesion area per histological section and overall infarct volume were markedly reduced in timp-3−/− compared to timp-3+/+ mice (Figure 7e and f). However, when the duration of MCAO was increased to 90 min, larger lesions occurred and no differences in striatal lesion volume could be discerned between the two strains (mean lesion volume of 74.0±37.7 versus 100.1±47.8 mm3 in timp-3+/+ versus timp-3−/− mice, mean±S.E.M., P=0.68, n=6 mice per strain). Collectively, these data suggest that timp-3−/− mice display increased resistance to mild transient focal ischemia, but are not protected against more severe lesions where necrotic death mechanisms are likely to predominate.

Lesion size in timp-3+/+ versus timp-3−/− at 3 days of reperfusion following 30 min transient MCAO. Timp-3+/+ and timp-3−/− mice were subjected to 30 min transient MCAO followed by 3 days of reperfusion. Coronal sections were stained for degenerating neurons using Fluoro-Jade (a, b) or stained for apoptotic nuclei using TUNEL (c, d). Infarct volume in timp-3+/+ versus timp-3−/− mice as assessed by morphometric analysis of Fluoro-Jade-stained histological sections, (e) n=5 mice per strain, P<0.001. (f) Average lesion area per histological section in rostrocaudal direction in timp-3+/+ versus timp-3−/− mice
Discussion
Our studies indicate that TIMP-3 modulates death receptor-mediated apoptosis of cortical neurons exposed to OGD in culture and modulates neuronal sensitivity to ischemia following mild, but not severe, in vivo MCAO. Exposure of cortical neurons to 2 h OGD results in a delayed apoptotic response that is dependent upon death receptor signaling and partially impaired in timp-3−/− cultures. The proapoptotic activity of TIMP-3 appears to be mediated through metalloproteinase inhibition, since resistance to OGD is reversed in timp-3−/− cultures by incubation with exogenous rTIMP-3 or the metalloproteinase inhibitor, GM6001. Disinhibition of metalloproteinase activity results in heightened shedding of the death ligand, FasL, in timp-3−/− cultures exposed to OGD, implicating FasL stabilization as one potential mechanism through which TIMP-3 promotes apoptosis in neurons. Finally, timp-3−/− mice display marked resistance to delayed neuronal cell death in a mild MCAO model of stroke. Taken together, these observations indicate that TIMP-3 is proapoptotic in neuronal ischemia, and that its effects are likely mediated through metalloproteinase inhibition and stabilization of cell-surface death receptors.
TIMP-3 is known to promote apoptosis in normal and malignant cells through increased death receptor signaling.20, 21, 22, 23 TIMP-3 overexpression sensitizes colon carcinoma cells to TNF-α-induced apoptosis via increased TNF receptor number on the cell surface.23 In vascular smooth muscle cells, TIMP-3 expression leads to apoptosis dependent on functional Fas-associated death domain and is inhibited by introduction of a dominant-negative FADD mutant.22 Exposure of melanoma cell lines to TIMP-3 (but not TIMP-1, -2 or -4) results in stabilization of three distinct death receptors, TNFR1, Fas and TRAILR1, and sensitizes these cells to apoptosis induced by their respective ligands both in vivo and in vitro.20 Overexpression of TIMP-3 in lung cancer cells results in apoptosis and increased p53, FasL, TNFR1 and TNFR2.31 The proapoptotic activity of TIMP-3 is dependent on metalloproteinase inhibition and has been mapped to the metalloproteinase inhibitory N-terminal domain of the protein.20, 32
Similarly, our studies suggest that TIMP-3 may promote neuronal apoptosis through stabilization of FasL following OGD in culture. In support of this, we found that TIMP-3 gene deletion results in the attenuation of receptor-mediated death, and that this phenotype is reversed by exogenous rTIMP-3 or by the broad-spectrum metalloproteinase inhibitor, GM6001. Also, we observed a marked increase in FasL release into the conditioned medium in timp-3−/− cultures exposed to OGD conditions, also reversed by the broad-spectrum metalloproteinase inhibitor, GM6001. It is well known that ectodomain shedding of FasL occurs through metalloproteinase activity at the cell surface,14, 16, 24 and that conversion of transmembrane FasL to the soluble form may diminish or enhance its death-promoting activity, depending on context.14, 16, 17, 24, 33 Similarly, we found that addition of soluble FasL was not toxic to our cultures, despite constitutive Fas and TIMP-3 expression. Previous reports have demonstrated that overexpression of transmembrane FasL,2 but not soluble Fas,2, 25 is quite effective at killing embryonic cortical neurons in culture. Thus, increased release of FasL from the cell surface could underlie the partial resistance to receptor-mediated death induced by OGD.
Our observations also suggest that TIMP-3 plays a proapoptotic role following cerebral ischemia in vivo. For these studies, we utilized a murine model of mild stroke, involving a brief 30 min transient MCAO, a model in which caspase-dependent delayed apoptotic death predominates.30 Under these conditions, the proapoptotic action of TIMP-3 was revealed, since timp-3−/− mice displayed reduced lesion size at 3 days. However, a more prolonged ischemic interval of 90 min masked these differences in susceptibility, which may be related to the fact that neuronal death following severe MCAO episodes is less susceptible to caspase inhibition and is likely to rely more on excitotoxic and necrotic mechanisms.30 Although the proapoptotic mechanisms of TIMP-3 in vivo have yet to be fully elucidated, the notion that TIMP-3 acts through stabilization of FasL is consistent with our in vitro findings and with previous work demonstrating TIMP-3 and FasL coexpression in TUNEL+ neurons within the penumbral region following MCAO.9
Our results do not exclude the role of other death receptors in ischemia and their regulation by TIMP-3. Of particular importance, TIMP-3 is a potent inhibitor of TACE (TNF-α-converting enzyme), which is a member of the ADAM gene family of metalloproteinases that cleaves TNF-α from the cell surface. Release of TNF-α from the cell surface by TACE can extend TNF effects from juxtacrine to paracrine, resulting in various biological effects dependent on cell and tissue context.34 Disinhibition of TACE has recently been shown to underlie enhanced inflammatory reactions in timp-3−/− mice subjected to a variety of physiological insults that target the vasculature,35 liver36 and systemic immune responses.37 In the CNS, TACE has been purported to play a neuroprotective role and to mediate ischemic tolerance after a preconditioning ischemic stimulus.38 It will be particularly important to ascertain TIMP-3 regulation of TACE in cerebral ischemia in future studies.
Recent studies on metalloproteinases in stroke indicate multiple roles, including regulation of blood brain barrier, proapoptotic and prosurvival functions (for review, see Cunningham et al.39). Metalloproteinase inhibitors have been purported to provide therapeutic benefit based on studies in rodents, due to the ability of these pharmacological inhibitors to reduce blood brain barrier damage at early times following stroke. Our studies provide a novel link between metalloproteinase activity and neuronal susceptibility to extrinsic death signals following OGD in the CNS and may have implications for the therapeutic treatment of stroke.
Materials and Methods
Primary cortical neuronal cultures
This study was approved by the University of New Mexico Animal Care and Use Committee and conformed to the NIH Guidelines for use of animals in research. Timed pregnant female mice were killed by halothane overdose and the embryos removed by cesarean section. Primary neuronal cultures were established from cerebral cortices of C57BL/6 timp-3+/+ and C57BL/6 timp-3−/−40 embryos at gestation day 15, using enzymatic dissociation with trypsin as previously described.27 For immunocytochemistry, 4.8 × 105 dissociated cells were plated on poly-L-lysine-coated coverslips (0.1 mg/ml; Sigma, St Louis, MO, USA) in 24-well plates. For biochemical procedures, 2.28 × 106 cells were plated on precoated poly-L-lysine six-well plates (BD Biosciences, San Diego, CA, USA). The cultures were maintained under serum-free conditions in neurobasal medium (Invitrogen Corp., Carlsbad, CA, USA), supplemented with B-27 supplement (2%; Invitrogen Corp.), glutamine (0.5 mM; Sigma), glutamate (25 μM; Sigma), penicillin (100 U/ml) and streptomycin (100 μg/ml; Invitrogen Corp.). At 4 days in vitro (DIV), half of the medium was removed and replaced with fresh medium without glutamate, as indicated by the manufacturer. The cultures were maintained in a humidified incubator at 37°C with 5% CO2. At 7 DIV, cultures were used for experimentation. The cultures were incubated with the following compounds: recombinant human TIMP-3 (1.25 μg/ml; generous gift from Dr. Gillian Murphy, Cambridge University), GM6001 (25 μM; Chemicon, Temecula, CA, USA), mouse Fas-Fc (5–20 μg/ml; R&D Systems Inc., Minneapolis, MN, USA), trimerized s-FasL (5–50 ng/ml; R&D Systems Inc.) or active catalytic domain of MMP-3 (10 or 100 μg/ml; Calbiochem).
Oxygen–glucose deprivation
Primary cortical neuronal cultures were placed in an anaerobic chamber (Coy Laboratories, Grass Lake, MI, USA) containing a gas mixture of 5% CO2, 5% H2 and 85% N2 (<0.2% O2). Normal culture media were replaced with deoxygenated, glucose-free Earle's balanced salt solution, and the cultures were maintained under glucose-free anaerobic conditions at 37°C for 2 h. OGD was terminated by returning the cultures to normoxic conditions and neurobasal medium supplemented with 2% B-27 supplement, 0.5 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. As a control, sister cultures were placed in Earle's balanced salt solution containing 25 mM glucose (Invitrogen Corp.) for 2 h at 37°C under normoxic conditions and then returned under normoxic conditions to neurobasal medium supplemented with 2% B-27 supplement, 0.5 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin.
Immunohistochemistry
Cortical cultures were fixed with 4% paraformaldehyde in 0.1 M sodium phosphate buffer and rinsed. The following antibodies were used at the indicated dilutions: rabbit polyclonal activated caspase-3 (1 : 100; Trevigen, Gaithersburg, MD, USA), mouse monoclonal anti-NeuN (1 : 100; Chemicon), polyclonal rabbit anti-TIMP-3 (1 : 1000; Chemicon), monoclonal mouse anti-MAP-2 (1 : 500; Sigma), polyclonal rabbit anti-FasL (1 : 500; Santa Cruz Biotechnology) and polyclonal rabbit anti-Fas (1 : 500; Santa Cruz Biotechnology). Immunofluorescence was visualized using either FITC- or CY3-conjugated secondary antibodies (1 : 250; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Cell nuclei were labeled with DAPI nuclear dye (300 nM). Quantitative analysis was performed by counting five random fields, blinded to the investigator, with a × 40 objective per coverslip using an upright Olympus microscope equipped with epifluorescence.
Western blotting
Total protein was separated by 15% SDS-PAGE and transferred to nitrocellulose immunoblotting membrane. The membranes were blocked with 190 BSA incubated with TIMP-3 primary antibody (1 : 500; Sigma) and immunoreactive bands were detected using donkey anti-rabbit secondary antibody conjugated to horseradish peroxidase (1 : 20 000; Jackson ImmunoResearch Laboratories). The femtoLucent chemiluminescence™ (Chemicon) protocol was used to detect immunoreactive bands according to the manufacturer.
Cell death ELISA
To detect and quantify histone-associated DNA fragments (mono- and oligonucleosomes) generated early in apoptosis and released into the cytoplasm, a photometric enzyme-linked immunoassay kit was used (Roche Applied Science, Mannheim, Germany). This assay detects apoptotic but not necrotic cell death. Cells or tissues were homogenized and the cytoplasmic fraction was isolated by the following centrifugations. The cell lysates were first spun at 750 × g for 10 min, the supernatants were collected and spun at 10 000 × g for 20 min, and finally the supernatants were again collected and spun 100 000 × g for 1 h at 4C. The final supernatant contained the cytoplasmic fraction. The ELISA was performed according to the manufacturer's instructions.
LDH assay
To detect the early loss of membrane integrity typical of necrosis, LDH was measured in conditioned media and total lysates of cortical cultures exposed to OGD for 2 h, using the Cytotox 96R Non-Radioactive Cytotoxicity Assay (Promega Corporation, Madison, WI, USA) according to the manufacturer's protocol. Bovine heart LDH equaling 10 000 lysed L929 fibroblast cells was used as a positive control.
FasL ELISA
To detect FasL present in cell culture conditioned medium, Quantikine® M mouse FasL immunoassay was used (R&D Systems Inc.). Cell culture medium was collected and concentrated 20-fold using Centricon YM-10 concentrators (Millipore Corp., Billerica, MA, USA). The FasL immunoassay was performed as indicated by the manufacturer's protocol.
Transient MCAO
Adult male timp-3+/+ and timp-3−/− mice were housed under a 12 h light:12 h dark cycle with food and water available ad libitum. Mice (20–25 g) were anesthetized with 4.0% halothane for induction of anesthesia and maintained on 1.0% halothane in 1 l O2 using a Fluotec 3 vaporizer (Ohio Medical Products, Madison, WI, USA). Body temperature was maintained at 37°C by a heating pad. The right common carotid artery was exposed through a midline incision. The external carotid artery was ligated with 6-0 silk suture. A 2-cm length of 6-0 rounded tip nylon suture was advanced from the common carotid through the internal carotid up to the level of the anterior cerebral artery. The suture was inserted 10–11 mm from the bifurcation of common carotid to occlude the middle cerebral artery. After 30 min of MCAO, the suture was slowly withdrawn to allow for reperfusion. Laser doppler (Moore Instruments Limited, Devon, UK) flowmetry was used monitor rCBF at 10 min before, 5 min during and 30 min after MCAO. All mice were subjected to behavioral tests prior to MCAO, during occlusion at 20 min and 30 min after the onset of reperfusion as described. Only mice that obtained a score ⩾2, as assessed by an observer blinded to surgical treatment, were used for experimentation. Mean arterial blood pressure was monitored in a subset of mice of each genotype 15 min before MCAO, during occlusion and 15 min after reperfusion using a blood pressure monitor system (Harvard Apparatus, Holliston, MA, USA). Blood was taken from the left femoral artery 15 min before MCAO and after perfusion. PO2, PCO2 and pH were quantified using Phox Basic blood gas analyzer (Nova Biomedical, Waltham, MA, USA). Temperature was continuously measured by rectal thermometer (Fisher, Pittsburgh, PA, USA).
Cerebral vasculature
To determine the patency of the Circle of Willis, normal adult male timp-3+/+ and timp-3−/− mice (n=5 each genotype) were anesthetized with pentobarbital (100 mg/ml) and transcardially perfused with 100 ml of 10 U/ml heparin in PBS followed by India Ink in an equal volume of 20% gelatin in H2O. The brain was removed, fixed in 4% paraformaldehyde for 24 h and the cerebral vasculature was observed with a dissecting microscope.
Fluoro-Jade staining and TUNEL
At 3 days following MCAO and reperfusion, mice were killed with sodium pentobarbital and immediately transcardially perfused with 2% paraformaldehyde containing 0.075 M lysine and 0.01 M sodium periodate. Brains were removed and cryoprotected in 30% sucrose in phosphate buffer and cryosectioned in the coronal plane at 16 μm thickness. Three consecutive sections were positioned per slide and the sets were stored at −80°C until use. For each analysis, every 10th slide was taken throughout the rostrocaudal extent of the brain, dehydrated, incubated with 0.06% potassium permanganate, rinsed in dH2O and transferred to 0.001% Fluoro-Jade (Histo-Chem Inc., Jefferson, AR, USA) in 0.1% acetic acid for 30 min with gentle agitation on ice. The sections were rinsed and coverslipped using Prolong AntiFade™ mounting media (Molecular Probes, Eugene, OR, USA). The average lesion area was quantified across all sections positive for Fluoro-Jade staining using Stereo Investigator software (Microbrightfield Inc, Williston, VT, USA). Lesion volume was determined by multiplying the average lesion area by thickness of the entire lesion. A separate histological set was used for TUNEL labeling, performed using NeuroTACS™ II kit (Trevigen) according to the manufacturer's protocol.
Statistics
Data are expressed as means±S.E.M. Significant differences between means were determined using analysis of variance with Tukey multiple comparisons post hoc analysis by Prism software (Graphpad Software, San Diego, CA, USA).
Abbreviations
- rTIMP-3:
-
recombinant tissue inhibitor of metalloproteinases-3
- OGD:
-
oxygen–glucose deprivation
- MCAO:
-
middle cerebral artery occlusion
- TNFR:
-
tumor necrosis factor receptor
References
Ashkenazi A, Dixit VM . Death receptors: signaling and modulation. Science 1998; 281: 1305–1308.
Qiu J, Whalen MJ, Lowenstein P, Fiskum G, Fahy B, Darwish R et al. Upregulation of the Fas receptor death-inducing signaling complex after traumatic brain injury in mice and humans. J Neurosci 2002; 22: 3504–3511.
Ethell DW, Buhler LA . Fas ligand-mediated apoptosis in degenerative disorders of the brain. J Clin Immunol 2003; 23: 439–446.
Hovelmeyer N, Hao Z, Kranidioti K, Kassiotis G, Buch T, Frommer F et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol 2005; 175: 5875–5884.
Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J et al. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ 2001; 8: 679–686.
Felderhoff-Mueser U, Taylor DL, Greenwood K, Kozma M, Stibenz D, Joashi UC et al. Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic–ischemic injury to the developing rat brain. Brain Pathol 2000; 10: 17–29.
Rosenbaum DM, Gupta G, D'Amore J, Singh M, Weidenheim K, Zhang H et al. Fas (CD95/APO-1) plays a role in the pathophysiology of focal cerebral ischemia. J Neurosci Res 2000; 61: 686–692.
Yew DT, Ping Li W, Liu WK . Fas and activated caspase 8 in normal, Alzheimer and multiple infarct brains. Neurosci Lett 2004; 367: 113–117.
Wallace JA, Alexander S, Estrada EY, Hines C, Cunningham LA, Rosenberg GA . Tissue inhibitor of metalloproteinase-3 is associated with neuronal death in reperfusion injury. J Cereb Blood Flow Metab 2002; 22: 1303–1310.
Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C et al. A critical role of neural-specific JNK for ischemic apoptosis. Proc Natl Acad Sci USA 2003; 100: 15184–15189.
Blankenberg FG, Kalinyak J, Liu L, Koike M, Cheng D, Goris ML et al. 99mTc-HYNIC-annexin V SPECT imaging of acute stroke and its response to neuroprotective therapy with anti-Fas ligand antibody. Eur J Nucl Med Mol Imaging 2006; 33: 566–574.
French LE, Tschopp J . Protein-based therapeutic approaches targeting death receptors. Cell Death Differ 2003; 10: 117–123.
Beier CP, Wischhusen J, Gleichmann M, Gerhardt E, Pekanovic A, Krueger A et al. FasL (CD95L/APO-1L) resistance of neurons mediated by phosphatidylinositol 3-kinase-Akt/protein kinase B-dependent expression of lifeguard/neuronal membrane protein 35. J Neurosci 2005; 25: 6765–6774.
Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stamenkovic I . Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res 2001; 61: 577–581.
Kheradmand F, Werb Z . Shedding light on sheddases: role in growth and development. BioEssays 2002; 24: 8–12.
Powell WC, Fingleton B, Wilson CL, Boothby M, Matrisian LM . The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr Biol 1999; 9: 1441–1447.
Tanaka M, Itai T, Adachi M, Nagata S . Downregulation of Fas ligand by shedding. Nat Med 1998; 4: 31–36.
Woessner Jr JF . That impish TIMP: the tissue inhibitor of metalloproteinases-3. J Clin Invest 2001; 108: 799–800.
Leco KJ, Khokha R, Pavloff N, Hawkes SP, Edwards DR . Tissue inhibitor of metalloproteinases-3 (TIMP-3) is an extracellular matrix-associated protein with a distinctive pattern of expression in mouse cells and tissues. J Biol Chem 1994; 269: 9352–9360.
Ahonen M, Poukkula M, Baker AH, Kashiwagi M, Nagase H, Eriksson JE et al. Tissue inhibitor of metalloproteinases-3 induces apoptosis in melanoma cells by stabilization of death receptors. Oncogene 2003; 22: 2121–2134.
Baker AH, George SJ, Zaltsman AB, Murphy G, Newby AC . Inhibition of invasion and induction of apoptotic cell death of cancer cell lines by overexpression of TIMP-3. Br J Cancer 1999; 79: 1347–1355.
Bond M, Murphy G, Bennett MR, Newby AC, Baker AH . Tissue inhibitor of metalloproteinase-3 induces a Fas-associated death domain-dependent type II apoptotic pathway. J Biol Chem 2002; 277: 13787–13795.
Smith MR, Kung H, Durum SK, Colburn NH, Sun Y . TIMP-3 induces cell death by stabilizing TNF-alpha receptors on the surface of human colon carcinoma cells. Cytokine 1997; 9: 770–780.
Mitsiades N, Poulaki V, Leone A, Tsokos M . Fas-mediated apoptosis in Ewing's sarcoma cell lines by metalloproteinase inhibitors. J Natl Cancer Inst 1999; 91: 1678–1684.
Wetzel M, Rosenberg GA, Cunningham LA . Tissue inhibitor of metalloproteinases-3 and matrix metalloproteinase-3 regulate neuronal sensitivity to doxorubicin-induced apoptosis. Eur J Neurosci 2003; 18: 1050–1060.
Jaworski DM, Fager N . Regulation of tissue inhibitor of metalloproteinase-3 (Timp-3) mRNA expression during rat CNS development. J Neurosci Res 2000; 61: 396–408.
Wetzel M, Tibbitts J, Rosenberg GA, Cunningham LA . Vulnerability of mouse cortical neurons to doxorubicin-induced apoptosis is strain-dependent and is correlated with mRNAs encoding Fas, Fas-Ligand, and metalloproteinases. Apoptosis 2004; 9: 649–656.
Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC et al. Caspase activation and neuroprotection in caspase-3-deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci USA 2002; 99: 15188–15193.
Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J et al. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci 1999; 19: 3809–3817.
Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T et al. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab 1998; 18: 238–247.
Finan KM, Hodge G, Reynolds AM, Hodge S, Holmes MD, Baker AH et al. In vitro susceptibility to the pro-apoptotic effects of TIMP-3 gene delivery translates to greater in vivo efficacy versus gene delivery for TIMPs-1 or -2. Lung Cancer 2006; 53: 273–284.
Bond M, Murphy G, Bennett MR, Amour A, Knauper V, Newby AC et al. Localization of the death domain of tissue inhibitor of metalloproteinase-3 to the N terminus. Metalloproteinase inhibition is associated with proapoptotic activity. J Biol Chem 2000; 275: 41358–41363.
Matsuno H, Yudoh K, Watanabe Y, Nakazawa F, Aono H, Kimura T . Stromelysin-1 (MMP-3) in synovial fluid of patients with rheumatoid arthritis has potential to cleave membrane bound Fas ligand. J Rheumatol 2001; 28: 22–28.
Georgopoulos S, Plows D, Kollias G . Transmembrane TNF is sufficient to induce localized tissue toxicity and chronic inflammatory arthritis in transgenic mice. J Inflamm 1996; 46: 86–97.
Kassiri Z, Oudit GY, Sanchez O, Dawood F, Mohammed FF, Nuttall RK et al. Combination of tumor necrosis factor-alpha ablation and matrix metalloproteinase inhibition prevents heart failure after pressure overload in tissue inhibitor of metalloproteinase-3 knock-out mice. Circ Res 2005; 97: 380–390.
Mohammed FF, Smookler DS, Taylor SE, Fingleton B, Kassiri Z, Sanchez OH et al. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet 2004; 36: 969–977.
Smookler DS, Mohammed FF, Kassiri Z, Duncan GS, Mak TW, Khokha R . Cutting edge: tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J Immunol 2006; 176: 721–725.
Pradillo JM, Romera C, Hurtado O, Cardenas A, Moro MA, Leza JC et al. TNFR1 upregulation mediates tolerance after brain ischemic preconditioning. J Cereb Blood Flow Metab 2005; 25: 193–203.
Cunningham LA, Wetzel M, Rosenberg GA . Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 2005; 50: 329–339.
Leco KJ, Waterhouse P, Sanchez OH, Gowing KL, Poole AR, Wakeham A et al. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3). J Clin Invest 2001; 108: 817–829.
Acknowledgements
This work was supported by NIH R01 NS04737 (LAC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by L Greene
Rights and permissions
About this article
Cite this article
Wetzel, M., Li, L., Harms, K. et al. Tissue inhibitor of metalloproteinases-3 facilitates Fas-mediated neuronal cell death following mild ischemia. Cell Death Differ 15, 143–151 (2008). https://doi.org/10.1038/sj.cdd.4402246
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402246
Keywords
This article is cited by
-
IGF1R+ Dental Pulp Stem Cells Enhanced Neuroplasticity in Hypoxia-Ischemia Model
Molecular Neurobiology (2017)
-
Role of IGF1R+ MSCs in modulating neuroplasticity via CXCR4 cross-interaction
Scientific Reports (2016)
-
SUMO-specific protease 1 protects neurons from apoptotic death during transient brain ischemia/reperfusion
Cell Death & Disease (2016)
-
Effect of Ermiao Recipe (二妙方) with medicinal guide Angelicae Pubescentis Radix on promoting the homing of bone marrow stem cells to treat cartilage damage in osteoarthritis rats
Chinese Journal of Integrative Medicine (2014)
-
Adaptive Changes in the Neuronal Proteome: Mitochondrial Energy Production, Endoplasmic Reticulum Stress, and Ribosomal Dysfunction in the Cellular Response to Metabolic Stress
Journal of Cerebral Blood Flow & Metabolism (2013)
