
Abstract
Accumulation of misfolded proteins in the endoplasmic reticulum (ER) induces the unfolded protein response (UPR), which alleviates protein overload in the secretory pathway. Although the UPR is activated under diverse pathological conditions, its physiological role during development and in adulthood has not been fully elucidated. Binding immunoglobulin protein (BiP) is an ER chaperone, which is central to ER function. We produced knock-in mice expressing a mutant BiP lacking the retrieval sequence to cause a defect in ER function without completely eliminating BiP. In embryonic fibroblasts, the UPR compensated for mutation of BiP. However, neonates expressing mutant BiP suffered respiratory failure due to impaired secretion of pulmonary surfactant by alveolar type II epithelial cells. Expression of surfactant protein (SP)-C was reduced and the lamellar body was malformed, indicating that BiP plays a critical role in the biosynthesis of pulmonary surfactant. Because pulmonary surfactant requires extensive post-translational processing in the secretory pathway, these findings suggest that in secretory cells, such as alveolar type II cells, the UPR is essential for managing the normal physiological ER protein overload that occurs during development. Moreover, failure of this adaptive mechanism may increase pulmonary susceptibility to environmental insults, such as hypoxia and ischemia, ultimately leading to neonatal respiratory failure.
Similar content being viewed by others


Main
Secretory proteins are subjected to quality control in the endoplasmic reticulum (ER) through interaction with molecular chaperones such as binding immunoglobulin protein (BiP), which functions as intermediaries for protein folding or degradation.1 Extracellular insults, such as ischemia, hypoxia, and genetic mutations, result in aberrant protein folding and accumulation of misfolded proteins in the ER. ER stress initiates the unfolded protein response (UPR), which enhances the capacity for ER quality control by reducing general protein synthesis, producing ER chaperones, and promoting ER-associated protein degradation (ERAD).2, 3 Failure of this adaptive mechanism can cause cellular dysfunction and cell death, resulting in diverse human disorders4, 5 such as neurodegenerative disease, cardiomyopathy,6 and diabetes.7
Respiratory distress syndrome in newborns is often associated with premature birth or low birth weight accompanied by reduced pulmonary surfactant production.8 Pulmonary surfactant is secreted by highly differentiated alveolar type II epithelial cells, and is composed of phospholipids and surfactant proteins (SP) A, B, C, and D. Surfactant reduces alveolar surface tension and keeps the alveolar space open, which is essential for lung function after the transition at birth from the embryonic fluid environment to air. SP-B and SP-C are small, highly hydrophobic proteins processed from proSP-B and proSP-C, respectively, during transport through the ER and the Golgi to the multivesicular body. Mature SP-B and SP-C are further transported to the lamellar body where they bind phospholipids before secretion into the alveolar space via regulated exocytosis. ProSP-B associated with BiP is found in the ER.9 Furthermore, mutations in SP-C cause the accumulation of misfolded SP-C in the ER, thereby activating the UPR,10, 11 resulting in interstitial lung disease in children and adults. This suggests that ER stress is involved in lung disease,12 but whether ER dysfunction causes lung disease remains an open question.
BiP, one of the most abundant ER chaperones, plays a central role in ER function, assisting in protein translocation, folding, degradation, and regulation of the UPR.13 ER chaperones are localized to the ER by two mechanisms — retention and retrieval.14 BiP is retained in the ER through interaction with other ER proteins and the ER matrix. When misfolded proteins accumulate in the ER, BiP dissociates from some ER membrane proteins, such as inositol-requiring kinase-1 (IRE1), PKR-like ER-associated kinase (PERK), and activating transcription factor 6 (ATF6). BiP dissociation activates these kinases and transcription factors and initiates the UPR,15 which leads to increased expression of X-box-binding protein-1 (XBP-1) and ATF4.3 When BiP is secreted from the ER along with misfolded proteins,16, 17 the C-terminal Lys-Asp-Glu-Leu (KDEL) sequence of BiP is recognized by the KDEL receptor, thereby facilitating the retrieval of BiP from post-ER compartments to the ER.18, 19 Yeast BiP (Kar2) is essential for survival; when the retrieval sequence (in yeast: His-Asp-Glu-Leu, HDEL) is deleted, a fraction of Kar2 is secreted from the ER. However, the UPR is activated and this maintains a minimal level of Kar2 in the ER.20
Complete depletion of BiP could be lethal for early embryonic cells.21 We therefore produced knock-in mice expressing a mutant BiP in which the retrieval sequence was deleted by homologous recombination. These mice were used to elucidate processes sensitive to ER stress during development and in adulthood. The mutant-BiP mice died several hours after birth. Analysis of neonatal mutants revealed that dedicated secretory systems essential for pulmonary development might include an adaptive mechanism whereby BiP function accommodates increased levels of secretory proteins.
Results
Constitutively active UPR compensates for loss of ER BiP in cultured mammalian cells
We used homologous recombination to produce knock-in mice expressing a mutant BiP lacking the C-terminal KDEL sequence (Figure 1). This mutant BiP contained a C-terminal HA tag. Mouse embryonic fibroblasts (MEFs) derived from homozygous BiP mutant embryos expressed mutant BiP instead of wild-type BiP but grew as well as wild-type MEFs. Mutant BiP localized to the ER, and its expression was enhanced by tunicamycin, which disrupts protein glycosylation in the ER, thereby inducing the UPR. These results are consistent with those for wild-type BiP and other ER chaperones containing a KDEL sequence (Figure 2a). However, in metabolic labeling experiments, a significant fraction of mutant BiP was found in the medium, reflecting deletion of the KDEL sequence and impaired retrieval of mutant BiP (Figure 2b). We estimate that under resting culture conditions, one-third of the newly synthesized mutant BiP was secreted. The remainder was retained in the ER. A fraction of wild-type BiP was also secreted into the medium during ER stress (tunicamycin treatment), indicating that retrieval by the KDEL receptor is saturable.

Generation of knock-in mice expressing a mutant BiP lacking the KDEL retrieval sequence. (a) Top, the wild-type allele containing the BiP gene. Exons are indicated as solid bars. Middle, the targeting vector for homologous recombination. Bottom, the recombinant allele. The external probes for Southern blot analysis are indicated by gray quadrangle. Xb, XbaI; H, HindIII; B, BamHI; Kp, KpnI; Xh, XhoI. (b) Southern blots of ES cell genomic DNA digested with XbaI. The 5′ external probe detected a 11.2-kb fragment in the wild-type allele and a 4.7-kb fragment in the mutant allele. The 3′ external probe detected a 11.2-kb fragment in the wild-type allele and a 8.0-kb fragment in the mutant allele. ‘+’ represents the wild-type allele, and ‘B’ represents the mutant allele. (c) Genotyping of mice by PCR

Deletion of the KDEL sequence impairs retrieval of mutant BiP. (a) MEFs from homozygous mutant (B/B) and wild-type (+/+) embryos treated with or without tunicamycin (Tm, 2.5 μg ml−1) for 12 h were double-stained with monoclonal anti-KDEL and polyclonal anti-HA and observed by confocal laser microscopy. The anti-KDEL recognizes BiP as well as other KDEL-containing proteins, such as GRP94. Scale bars represent 10 μm. (b) Tm-treated (2.5 μg ml−1 for 12 h) or untreated MEFs were subjected to pulse-chase (p and c) labeling with[35S]methionine. Proteins in cell lysates (p and c) and in the culture medium (m) were immunoprecipitated with anti-KDEL or anti-HA. The proportion of secreted mutant BiP (m; 0.37) was evaluated by densitometry
Tunicamycin sensitivity and expression of mutant BiP was also confirmed by Western blotting (Figure 3a and b). Both mutant BiP and wild-type BiP were recognized by an antibody against the N-terminus of BiP. However, anti-KDEL only recognized wild-type BiP and GRP94, an ER chaperone with the KDEL sequence. Basal expression of XBP1, ATF4, phospho-PERK, and another ER chaperone, calreticulin, was enhanced in homozygous mutant BiP MEFs. Basal expression of mutant BiP mRNA was also enhanced in mutant MEFs (Figure 3c), indicating that the UPR was constitutively active. Thus, as seen previously in yeast,20 constitutive UPR activation maintains a minimal level of BiP (mutant or wild type) in the ER of mammalian cells, thus compensating for deletion of the KDEL sequence.

Loss of wild-type BiP is compensated by constitutive activation of the UPR in mammalian cells. (a) Tunicamycin (Tm)-treated (2.5 μg ml−1 for 12 h) or untreated MEFs from homozygous (B/B), heterozygous (B/+) and wild-type (+/+) embryos were collected. Expression of BiP, GRP94, mutant BiP, calreticulin, XBP1, ATF4, and γ-tubulin was determined by Western blotting. (b) Tm-treated (2.5 μg ml−1 for 0, 3, 6, 12, and 24 h) MEFs from homozygous (B/B) and wild-type (+/+) embryos were collected. Expression of BiP, GRP94, mutant BiP, phospho-PERK, and γ-tubulin was determined by Western blotting. The expression of each protein was normalized to that of γ-tubulin. (c) Northern blot of BiP mRNA expression in MEFs from homozygous (B/B) and wild-type (+/+) embryos treated with Tm (2.5 μg ml−1) at 37°C for 0–6 h. The expression of BiP mRNA was normalized to that of β-actin mRNA
Mutant-BiP embryos die shortly after birth
BiP was ubiquitously expressed in both mutant and wild-type embryos (Figure 4a and b). In all tissues examined, GRP94 expression was greater in homozygous BiP mutant embryos than in wild-type embryos (Figure 4b), suggesting that homozygous BiP mutant mice may suffer from global ER stress. Homozygous mutant-BiP embryos weighed less than wild types and heterozygotes at embryonic day (E) 18.5 (Figure 4c). Homozygous BiP mutant mice, shown in Figure 4d, were born at the expected Mendelian ratio of 1 :2: 1 (84 :182 :90, wild type :heterozygous :homozygous). Neonatal BiP mutants moved well and responded to painful stimuli, but they appeared pale and cyanotic. They also cried less and displayed shallow breathing. The neonatal homozygous mutants generally died within several hours of birth; thus, we suspected that the observed lethality might reflect respiratory problems.

Mutant-BiP embryos die shortly after birth. (a) BiP expression was ubiquitous in both mutant and wild-type embryos. Whole-mount in situ hybridization revealed ubiquitous expression of BiP and mutant BiP in E9.5 embryos. Scale bar represents 200 μm. (b) Western blots of BiP or mutant-BiP in tissues from wild type (+) and homozygous mutant (b) E18.5 embryos, respectively. Expression of GRP94 was normalized to that of γ-tubulin. In all tissues examined, GRP94 was greater in mutants than in wild type. (c) Body weights (mean±S.D.) of homozygous (B/B, n=14), heterozygous (B/+, n=23) and wild-type (+/+, n=15) E18.5 embryos. Homozygous mutant embryos weighed significantly less than wild types or heterozygotes (P<0.0001). (d) Gross appearance of wild type (+/+) and homozygous (B/B) P1 neonates. (e) Lungs from E18.5 mice delivered by Cesarian section
When delivered by Caesarian section at E18.5 and killed before breathing, gross morphology of the lungs (Figure 4e) and airways from BiP mutant mice was indistinguishable from wild type (Figure 5a). Wild-type and homozygous BiP mutant embryonic alveoli had an equivalent distribution of alveolar type II cells expressing the SP-C (Figure 5b). However, histological examination of lungs isolated from neonatal BiP mutants several hours after birth revealed atelectasis with poor inflation of peripheral airways. Hemorrhage and cell debris were also observed in the mutant alveolar space. Alveolar epithelia in BiP mutant mice were enlarged, whereas, as expected, those in wild-type neonates were distended (Figure 5c). These observations indicate that homozygous neonatal BiP mutants developed atelectasis and respiratory failure after birth.

The gross morphology of lungs and airways from BiP mutant embryos is indistinguishable from wild type. (a, b) Lungs from E18.5 mice delivered by Cesarian section. (a) Sections stained with hematoxylin and eosin. +/+: wild-type, B/B: homozygous mutant. (b) Alveolar type II epithelial cells were stained with anti-SP-C (brown). Nuclei were stained with hematoxylin (violet). (c) Lung sections from P1 neonates. Atelectasis and cell debris were evident in the peripheral airways of neonatal mutants. +/+: Wild-type, B/B: homozygous mutant. (d) Lungs from a P1 neonatal BiP mutant (B/B) administered 50 μl of perfluorocarbon via the oropharynx and treated with 40% oxygen for 6 h. Sections were stained with hematoxylin and eosin. Scale bars represent 200 and 20 μm in the low- and high-magnification images, respectively
Altered biosynthesis of SPs in mutant alveolar type II cells
To examine whether a deficiency of pulmonary surfactant contributes to respiratory failure in homozygous BiP mutant mice, perfluorocarbon, a substitute for pulmonary surfactant, was administered into the oropharynx. Perfluorocarbon with oxygen treatment improved the activity of neonatal BiP mutants, turned their skin color from pale to pink, and improved lung inflation (Figure 5d). Expression of SPs in neonatal lung was examined by Western blotting (Figure 6a). Expression of SP-A and, more prominently, proSP-C, was reduced in mutant lungs compared with wild type, but there was no significant difference in proSP-B and SP-D expression. RT-PCR analysis revealed that the marked reduction of proSP-C in neonatal mutant lung was not due to reduced transcription (Figure 6b). Importantly, after birth, the expression of proSP-C was enhanced only in wild-type neonates (Figure 6c), suggesting that proSP-C might be degraded post-translationally in neonatal type II cells from BiP mutants. Furthermore, expression of CHOP, a transcription factor related to cell death during ER stress,22 increased in homozygous mutant lungs after birth, suggesting that mutant lung tissue might be suffering from ER stress. Heterozygous BiP mutants were viable and grew to adulthood. Furthermore, heterozygous expression of wild-type BiP sustained SP-C expression and suppressed CHOP expression in lung (Figure 6d and e), suggesting that the wild-type BiP is essential for SP-C biosynthesis.

Biosynthesis of pulmonary surfactant is impaired in mutant-BiP lung. (a) Western blots of SP-A, SP-B (proSP-B), SP-C (proSP-C), SP-D and γ-tubulin in lungs from P1 wild types (+/+) and mutants (B/B). The expression of each protein was normalized to that of γ-tubulin. (b) SP-C mRNA in lungs from P1 wild-types (+/+) and mutants (B/B) was evaluated by semi-quantitative RT-PCR. Serial dilutions of cDNA were standardized to GAPDH. (c) Western blots of pro-SP-C, GRP94, BiP, mutant BiP, CHOP, and γ-tubulin in lungs from E18.5 (E) and P1 (P) mice. (d) Western blots of SP-C (proSP-C) and γ-tubulin in lungs from P1 wild-type (+/+), heterozygous (B/+), and homozygous (B/B) mutant mice. The expression of SP-C was normalized to that of γ-tubulin. (e) Western blots of mutant BiP, CHOP, and γ-tubulin in P1 lungs
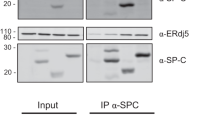
The subcellular localization of SP-A and SP-C was evaluated by confocal laser microscopy. In wild-type neonatal alveolar type II cells, SP-A and SP-C (proSP-C) colocalized with BiP, and other KDEL sequence–containing ER chaperones, in the ER. SP-A accumulated in the alveolar lining area of BiP mutant mice and costained with mutant BiP (Figure 7a). By contrast, SP-C remained in the ER, and its expression was markedly reduced in type II cells of neonatal BiP mutants (Figure 7b). Mature SP-B and SP-C are transported to the lamellar body where they bind phospholipids and are then secreted into the alveolar space via regulated exocytosis, whereas SP-A and SP-D are secreted independently of the lamellar body.12 Together, these data suggest that mutant BiP impairs the secretion of pulmonary surfactant, especially secretion through the lamellar body.

SP-A and SP-C in lungs from BiP mutants. (a) Costaining of lungs from P1 wild-type (+/+) and mutant (B/B) mice with anti-SP-A (brown staining, right panels) and monoclonal anti-KDEL or anti-HA (shown in green in different panels). (b) Costaining of lungs from P1 wild types (+/+) and mutants (B/B) with anti-SP-C (proSP-C; right panels) and anti-KDEL or anti-HA. Arrows indicate colocalization of SP-A or SP-C with KDEL-containing chaperones in wild-type type II cells as well as colocalization of SP-A with mutant BiP in B/B type II cells
Embryonic type II cells store glycogen in the cytoplasm, and this glycogen is consumed as the synthesis of pulmonary surfactant expands after birth. Type II cells in neonatal BiP mutants contained vacuole structures. Periodic acid Schiff (PAS) staining revealed cytoplasmic polysaccharides in these cells, even after birth (Figure 8a). Ultrastructural analysis of type II cells from neonates confirmed that cytoplasmic glycogen was indeed still present in mutant, but not in wild type, cells (Figure 8b and c). More importantly, the structure of the lamellar body was abnormal in embryonic and neonatal mutant type II cells. The lamellar body in wild-type neonates had wavy, dense laminations with clefting, whereas in BiP mutant neonates the lamellar body had loosely formed lamellar structures or was almost empty. These results indicate that the biosynthesis and secretion of pulmonary surfactant was impaired in BiP mutant type II cells.

Defective lamellar body formation in neonatal mutant-BiP alveolar type II cells. (a) Neonatal mutant type II cells contained vacuole structures. Alveolar epithelium from P1 wild type (+/+) and mutant (B/B) mice stained with toluidine blue (upper panels) or PAS (lower panels). Scale bars represent 10 μm. (b, c) Ultrastructure of the alveolar epithelium from wild type (+/+) and mutant-BiP (B/B) P1 neonates (b) and E18.5 embryos (c). Scale bars represent 1 μm. Arrows indicate lamellar bodies in type II cells. Cytoplasmic glycogen (gl) remained in neonatal mutant type II cells
SP-C is a small, highly hydrophobic protein processed from proSP-C during its transport through the ER and the Golgi to the multivesicular body. Mature SP-C is transported further to the lamellar body where it binds phospholipids before secretion into alveolar space via regulated exocytosis. Lamellar body formation is defective in alveolar type II cells of neonatal BiP mutants. Therefore, SP-C may be degraded by endosomal/lysosomal degradation and/or the ERAD pathway. Punctate SP-C is colocalized with KDEL-containing ER chaperones in both wild-type and homozygous mutant type II cells (Figure 9a), suggestive of ER accumulation of SP-C. The fraction of ER accumulation of SP-C in the mutant type II cells was 0.84, while that of Golgi accumulation was 0.08, evaluated by confocal colocalization images (Figure 9a and b).

SP-C accumulation in the ER. (a) Lungs from a P1 wild-type neonate (+/+) and a homozygous mutant (B/B) costained with polyclonal anti-SP-C and monoclonal anti-KDEL (ER staining). Colocalization of SP-C with KDEL-containing ER chaperones was seen in both wild type and mutant alveolar type II cells. (b) Lungs from a P1 wild-type neonate (+/+) and a homozygous mutant (B/B) co-stained with polyclonal anti-SP-C and monoclonal anti-giantin (Golgi complex staining). Scale bar represents 10 μm. The fractions of ER-localizing SP-C and Golgi-localizing SP-C were calculated by confocal colocalization images. ER-localizing SP-C to total SP-C in wild type; 0.81±0.052, Golgi-localizing SP-C in wild type; 0.12±0.032, ER-localizing SP-C in mutant; 0.84±0.036 (significantly more than that of wild types by t-test, P<0.018), Golgi-localizing SP-C in mutant; 0.08±0.043 (significantly less than that of wild types, P<0.0015). n=20, mean±S.D. (c, d) Inhibition of proteasomal degradation promotes ER accumulation of SP-C in homozygous mutant MEFs. MEFs from wild type (+/+, c) and homozygous mutant (B/B, d) embryos were transfected with SP-C with or without ALLN (10 μg ml−1; 12 h) then double-stained with monoclonal anti-KDEL (red) and polyclonal anti-SP-C (green). Anti-KDEL recognizes BiP as well as other KDEL-containing proteins, such as GRP94. Scale bars represent 10 μm
To examine the effect of proteasome inhibitors on SP-C, MEFs from wild-type and homozygous mutant embryos were transfected with SP-C in the presence or absence of a proteasome inhibitor (Figure 9c and d). Although the proteasome inhibitor enhanced SP-C expression in wild-type and BiP mutant MEFs, it enhanced surface expression of SP-C in wild-type MEFs but promoted ER accumulation of SP-C in the BiP mutant MEFs. Although endosomal/lysosomal degradation of SP-C cannot be excluded, these data suggest that, in homozygous mutant-BiP type II cells, misfolded SP-C may accumulate in the ER and be degraded by the ERAD pathway.
Discussion
We produced knock-in mice expressing a mutant BiP lacking the retrieval sequence to examine the effects of defects in the secretory pathway stress response without completely eliminating BiP function—as would be the case in BiP knockout mice.21 Mutant BiP predominantly affected dedicated secretory cells, such as alveolar type II cells, in which active secretion is particularly important. Putative impairment of protein folding in these mutant cells probably caused the observed respiratory failure and high neonatal mortality.
Deletion of the retrieval sequence from BiP, and the consequent lack of mutant-BiP recycling, could have two possible effects. First, the folding environment in the ER may be impaired. However, mutant BiP is functional as long as it remains in the ER. Therefore, constitutive activation of the UPR could compensate for the altered folding environment by producing mutant BiP in quantities sufficient for cell survival. Second, quality control in post-ER compartments may be affected. Proper ER-to-Golgi transport and subsequent ER retrieval of proteins and lipids is thought to contribute to quality control.16, 17, 23 In this regard, the folding (and therefore function) of pulmonary SPs, especially proSP-C, may depend on proper ER retrieval of BiP via the KDEL receptor.
Respiratory distress syndrome of newborns, also called hyaline membrane disease, causes high mortality and often accompanies pre-term delivery or low birth weight with reduced expression of pulmonary surfactant. Surfactant therapy combined with mechanical ventilation and other intensive care measures has reduced the mortality rate of this syndrome to below 10%.24 Surfactant proteins are required for proper lung development and function. Respiratory diseases result in humans and other mammals when these proteins are lacking. SP-B is critical for lung surfactant formation and function, and its deficiency causes immediate and severe neonatal respiratory failure in humans and mice.25, 26 Deletion of SP-B induces aberrant processing of proSP-C as well as defects in lamellar body formation. SP-C deficits are related to acute and chronic infant lung diseases in humans27 and respiratory failure in cattle.28 Furthermore, mutations in proSP-C have been correlated with chronic interstitial pneumonia.11, 12 ProSP-C is an type II integral membrane protein with structural homology to the amyloidogenic BRI family of proteins, which cause neurodegenerative dementia.29 Mutant proSP-C tends to misfold and may cause protein aggregation and ER stress.12 Thus, in the present study, aberrant quality control in the mutant-BiP type II epithelial cells may have resulted in proSP-C misfolding. Misfolded proSP-C may act in concert with decreased pulmonary surfactant levels to generate respiratory failure by causing ER stress in mutant type II cells.
Mutant-BiP mice have a distinct phenotype, as is the case for mice lacking other ER molecular chaperones. Hsp47 is responsible for collagen biosynthesis, and Hsp47 knockout mice die on E11.5.30 Calreticulin and calnexin participate in glycoprotein folding in the ER. Calreticulin knockout mice are embryonic lethal and display defective cardiac development.31 Calnexin knockout mice die during the early postnatal period, between birth and 3 months of age. These mice exhibit motor disorders owing to a loss of large myelinated nerve fibers.32
The UPR is a ubiquitous mechanism for adapting to ER stress, and BiP is an essential component of this system. However, at various developmental stages, some cell types require specific UPR signaling systems33 and chaperones. Mutant mouse models have revealed that the UPR plays a vital role during development by increasing protein synthesis as needed in dedicated secretory cells,34 such as pancreatic β cells,35 plasma cells36 and hepatocytes.37 Inadequate adaptation to these types of physiological demands may lead to diverse diseases.
Diminished quality control in the ER and in post-ER compartments of the mutant-BiP alveolar type II cells may be sufficient embryonically but not neonatally when increased biosynthesis of proSP-C and other SPs requires sufficient folding capacity. Indeed, at E18.5, proSP-C expression in type II cells was equivalent in lungs from BiP mutants and wild types (Figures 5b and 6c). After birth, proSP-C expression increased markedly in wild-type lungs but not in mutant lungs. SP-C mRNA level was, however, preserved in the BiP mutants, suggesting that proSP-C may be degraded co- or post-translationally in neonatal mutants.
Our results suggest that in neonates a physiological increase in SP production causes ER stress in dedicated secretory cells, like alveolar type II epithelial cells. This may increase susceptibility to environmental insults such as hypoxia and ischemia, leading to respiratory failure due to the loss of pulmonary surfactant and accumulation of misfolded proteins in the ER.
Materials and Methods
Cells, reagents, and general procedures
MEFs were prepared from E13.5 embryos. MEFs were grown at 37°C in an atmosphere of 5% CO2 in complete medium consisting of Dulbecco’s modified essential medium (DMEM; Sigma Chemical Co.) with 15% fetal bovine serum (FBS), 2 mM glutamine, 50 μg ml−1 streptomycin and 50 U ml−1 penicillin G.
The following antibodies were used: rabbit antiserum against calreticulin (Affinity Bioreagent), rabbit antiserum against XBP-1, rabbit antiserum against ATF4, rabbit antiserum against CHOP/GADD153, goat polyclonal antiserum against BiP/GRP78, rabbit antiserum against SP-A (H-148), goat polyclonal antiserum against SP-B (R-19), rabbit antiserum against SP-C (FL-197), rabbit antiserum against SP-D (C-18), rabbit antiserum against ubiquitin (all from Santa Cruz Biotechnology), rabbit anti-serum against the HA epitope (Zymed), mouse monoclonal antibody (mAb) against γ-tubulin (Sigma Chemical), mouse mAb SPA-827 against BiP (KDEL sequence; Stressgen), mouse mAb G1/133 against giantin (Alexis Biochemicals), Cy-2- or Cy-3-conjugated donkey antibody against rabbit IgG, and Cy-2- or Cy-3-conjugated donkey antibody against mouse IgG (Jackson Immunoresearch Laboratories). Tunicamycin was purchased from Nacali Tasque. Perfluorocarbon (perfluoro-2-butyltetrahydrofuran) was purchased from Fluorochem. N-acetyl-l-leucinal-l-lecinal-l-norleucinal (ALLN) was purchased from Sigma Chemical.
Metabolic labeling experiments and Western blotting were performed as described previously.6 Densitometry was performed using BAS 2500 and Image Gauge software (Fuji Photo Film Co., Ltd.) for metabolic labeling experiments and using LAS 1000 (Fuji Photo Film Co., Ltd.) and ImageJ software (Wayne Rasband, NIH) for Western blotting.
Generating mutant-BiP mice
All animal experimental procedures were in accordance with a protocol approved by the Institutional Animal Care Committee of Chiba University, Chiba, Japan. A rat BiP cDNA was used as a probe to isolate a genomic clone containing the whole exon of the BiP gene38 from the 129/SvJ mouse genomic library in λFIXII (Stratagene). A 0.6-kb BamHI/XhoI fragment encoding the C-terminal end of BiP and the stop codon, but lacking the KDEL sequence, was obtained by PCR. Then, part of a 2.1-kb genomic fragment containing the last three exons was replaced by the 0.6-kb fragment. The resultant fragment was used as a short arm for the targeting vector. A 0.5-kb XbaI/KpnI fragment, encoding the 3′ untranslated region of the exon after the stop codon and the 2.5-kb intron that followed, was amplified by a PCR and used as a long arm for the targeting vector. An XhoI/XbaI fragment containing a neomycin selection cassette flanked by loxP sequences (PHR68, a gift from T Kondo, Saitama, Japan) and a 2.7-kb fragment containing the HSV-thymidine kinase gene were introduced into the targeting vector to allow negative and positive selection. The targeting vector was linearized with NotI digestion and used for electroporation into R1 ES cells. The mutant allele resulting from homologous recombination had artificial XhoI and XbaI sites. Genomic DNA from G418-resistant clones (G418 was from Life Technologies) was digested with XbaI and analyzed by Southern blotting using an XbaI/HindIII fragment for the 5′-end probe and a PCR-amplified fragment for the 3′-end probe, as shown in Figure 1. Several homologous recombinants were obtained, and germline chimeras were generated as described.39 Resulting male chimeras were mated to C57BL/6 females. Tail DNA from the offspring was screened by PCR using the following oligonucleotides: 5′-gatcagtgcacctacaactc-3′ and 5′-gctcaaagagcgcattgac-3′ for the wild-type allele, and 5′-gtgaacgacccctaacaaaa-3′ and 5′-agcgtaatctggaacatcgt-3′ for the recombinant allele. To remove the neomycin selection cassette, heterozygous mice were mated with mice expressing Cre under the control of a CAG promoter. Heterozygous mice lacking neomycin were interbred to obtain homozygous mutant mice.
Detection of mRNA
Northern blot analysis was carried out as described.6 Expression of BiP and β-actin mRNAs was evaluated using cDNA probes encoding rat BiP (a gift from Dr. HRB Pelham) and mouse β-actin. Radioactivity was measured using BAS 2500 and Image Gauge software (Fuji Photo Film Co. Ltd.). The expression of BiP mRNA was normalized to that of β-actin. RT-PCR analysis was performed with the following primers: 5′-acctcaaacgccttctcatc-3′ and 5′-tttctgggcaggagcagctt-3′ for SP-C, and 5′-atggggtgaggccggtgctg-3′ and 5′-cttgatgtcatcatacttgg-3′ for GAPDH.
Whole-mount in situ hybridization
Whole-mount in situ hybridization was performed using digoxigenin-labeled riboprobes as described40 with rat BiP cDNA.
Transfection
A cDNA encoding mouse SP-C was obtained from wild-type lung mRNA using the following primers: 5′-ccttacaaaatggacatgag-3′ and 5′-cggatccacgatatagtagagtggtagct-3′. The cDNA was subcloned into a pcDNA3.1 myc-His vector (Invitrogen). The DNA sequence was verified using the Applied Biosystems ABI Prism 310 genetic analyzer. Transfection was performed with Fugene 6 (Roche).
Histochemistry
Lungs were isolated and fixed in 4% paraformaldehyde for 24 h. After fixation, they were dehydrated in increasing concentrations of ethanol and embedded in paraffin wax. Sections (8 m) were stained with hematoxylin and eosin. For immunohistochemistry, sections were incubated with 10% normal goat or bovine serum in phosphate-buffered saline (PBS) for 30 min to block non-specific antibody binding, and then incubated with a primary antibody in PBS for 12 h at 4°C. Sections were rinsed with PBS, incubated with a secondary antibody in PBS for 2 h at room temperature and then visualized using the VECTASTAIN Elite ABC kit (Vector Laboratories) with diaminobenzidine (Sigma).
Confocal and immunofluorescence microscopy
Cells on coverslips were fixed in 2% formaldehyde in PBS for 10 min at room temperature and then processed as described.6 Labeled cells were examined using a confocal laser scanning microscope (Axiovert 100M, LSM510, Ver. 3.2, Carl Zeiss) fitted with krypton and argon lasers and a Plan-Apochromat 100 NA 1.40 oil immersion objective.
Electron microscopy
Fifteen newborn animals and 24 embryos were processed for electron microscopy. Newborn animals were fixed within 2 h of birth. Embryos were fixed on E18.5. Before fixation, a tail sample was collected for genotyping.
Animals were anesthetized on ice. The thorax was opened, and the heart was removed with part of the large vessels. The trachea was clamped with tweezers, and both the left and right lobes of the lung were excised after cutting the trachea just proximal to the clamped site. The lung was immediately placed in a syringe filled with 3% glutaraldehyde in 10 mM HEPES, 145 mM NaCl (pH 7.4). After the air in the lung and air ducts was expelled by manipulating the plunger, the lung was cut into small pieces with a razor blade and was further fixed in the same fixative for an additional 2 h. After postfixation with 1% osmium tetroxide, the tissue was dehydrated in absolute ethanol and embedded in Epon mixture (TAAB, Berks, England). Ultra-thin sections, double-stained with uranyl acetate and lead citrate, were observed using an electron microscope (1200EX-II, JEOL, Tokyo, Japan).
Statistical analysis
Statistical analyses were performed by one-way analysis of variance and the Bonferroni test and t-test using KaleidaGraph software (Synergy Software, Reading, PA, USA).
Abbreviations
- BiP:
-
binding immunoglobulin protein
- ER:
-
endoplasmic reticulum
- ERAD:
-
ER-associated protein degradation
- MEF:
-
mouse embryonic fibroblast
- PAS:
-
periodic acid Schiff
- SP:
-
surfactant protein
- UPR:
-
unfolded protein response
References
Ellgaard L, Helenius A . Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 2003; 4: 181–191.
Patil C, Walter P . Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 2001; 13: 349–355.
Schroder M, Kaufman RJ . The Mammalian unfolded protein response. Annu Rev Biochem 2005; 74: 739–789.
Kopito RR, Ron D . Conformational disease. Nat Cell Biol 2000; 2: E207–E209.
Kaufman RJ . Orchestrating the unfolded protein response in health and disease. J Clin Invest 2002; 110: 1389–1398.
Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y et al. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol 2004; 24: 8007–8017.
Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 2002; 109: 525–532.
Clark H, Clark LS . The genetics of neonatal respiratory disease. Semin Fetal Neonatal Med 2005; 10: 271–282.
Korimilli A, Gonzales LW, Guttentag SH . Intracellular localization of processing events in human surfactant protein B biosynthesis. J Biol Chem 2000; 275: 8672–8679.
Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF . A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol 2005; 32: 521–530.
Bridges JP, Xu Y, Na CL, Wong HR, Weaver TE . Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP-C. J Cell Biol 2006; 172: 395–407.
Beers MF, Mulugeta S . Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol 2005; 67: 663–696.
Hendershot LM . The ER function BiP is a master regulator of ER function. Mt Sinai J Med 2004; 71: 289–297.
Sonnichsen B, Fullekrug J, Nguyen Van P, Diekmann W, Robinson DG, Mieskes G . Retention and retrieval: both mechanisms cooperate to maintain calreticulin in the endoplasmic reticulum. J Cell Sci 1994; 107: 2705–2717.
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D . Dynamic interaction of BiP and ER stress transducers in the unfolded- protein response. Nat Cell Biol 2000; 2: 326–332.
Hammond C, Helenius A . Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol 1994; 126: 41–52.
Yamamoto K, Fujii R, Toyofuku Y, Saito T, Koseki H, Hsu VW et al. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J 2001; 20: 3082–3091.
Munro S, Pelham HR . A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987; 48: 899–907.
Lewis MJ, Pelham HR . A human homologue of the yeast HDEL receptor. Nature 1990; 348: 162–163.
Beh CT, Rose MD . Two redundant systems maintain levels of resident proteins within the yeast endoplasmic reticulum. Proc Natl Acad Sci USA 1995; 92: 9820–9823.
Luo S, Mao C, Lee B, Lee AS . GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol 2006; 26: 5688–5697.
Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 1998; 12: 982–995.
Taxis C, Vogel F, Wolf DH . ER-golgi traffic is a prerequisite for efficient ER degradation. Mol Biol Cell 2002; 13: 1806–1818.
Hallman M . Lung surfactant, respiratory failure, and genes. N Engl J Med 2004; 350: 1278–1280.
Nogee LM, de Mello DE, Dehner LP, Colten HR . Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med 1993; 328: 406–410.
Clark JC, Wert SE, Bachurski CJ, Stahlman MT, Stripp BR, Weaver TE et al. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc Natl Acad Sci USA 1995; 92: 7794–7798.
Lahti M, Marttila R, Hallman M . Surfactant protein C gene variation in the Finnish population – association with perinatal respiratory disease. Eur J Hum Genet 2004; 12: 312–320.
Danlois F, Zaltash S, Johansson J, Robertson B, Haagsman HP, van Eijk M et al. Very low surfactant protein C contents in newborn Belgian White and Blue calves with respiratory distress syndrome. Biochem J 2000; 351 (Part 3): 779–787.
Kim SH, Creemers JW, Chu S, Thinakaran G, Sisodia SS . Proteolytic processing of familial British dementia-associated BRI variants: evidence for enhanced intracellular accumulation of amyloidogenic peptides. J Biol Chem 2002; 277: 1872–1877.
Nagai N, Hosokawa M, Itohara S, Adachi E, Matsushita T, Hosokawa N et al. Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J Cell Biol 2000; 150: 1499–1506.
Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH et al. Calreticulin is essential for cardiac development. J Cell Biol 1999; 144: 857–868.
Denzel A, Molinari M, Trigueros C, Martin JE, Velmurgan S, Brown S et al. Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol Cell Biol 2002; 22: 7398–7404.
Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT . Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006; 124: 587–599.
Wu J, Kaufman RJ . From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ 2006; 13: 374–384.
Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P . Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 2001; 7: 1165–1176.
Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM . Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001; 412: 300–307.
Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J . An essential role in liver development for transcription factor XBP-1. Genes Dev 2000; 14: 152–157.
Haas IG . and Meo T cDNA cloning of the immunoglobulin heavy chain binding protein. Proc Natl Acad Sci USA 1988; 85: 2250–2254.
Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC . Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA 1993; 90: 8424–8428.
Wilkinson DG, Nieto MA . Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. Methods Enzymol 1993; 225: 361–373.
Acknowledgements
We thank Dr. T Nishino for critical comments. We also thank R Kimura and R Fujii for excellent technical assistance. This work was supported by Grants-in-Aid for Science Research from the Ministry of Education, Culture, Sports, Science and Technology to TA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Piacentini
Rights and permissions
About this article
Cite this article
Mimura, N., Hamada, H., Kashio, M. et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ 14, 1475–1485 (2007). https://doi.org/10.1038/sj.cdd.4402151
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402151
Keywords
This article is cited by
-
Involvement of the unfolded protein response in the protective effects of growth hormone releasing hormone antagonists in the lungs
Journal of Cell Communication and Signaling (2021)
-
Secretion of the endoplasmic reticulum stress protein, GRP78, into the BALF is increased in cigarette smokers
Respiratory Research (2017)
-
Sublethal endoplasmic reticulum stress caused by the mutation of immunoglobulin heavy chain-binding protein induces the synthesis of a mitochondrial protein, pyrroline-5-carboxylate reductase 1
Cell Stress and Chaperones (2017)
-
The Role of Endoplasmic Reticulum Stress in Diabetic Nephropathy
Current Diabetes Reports (2017)
-
Differential roles of NF-Y transcription factor in ER chaperone expression and neuronal maintenance in the CNS
Scientific Reports (2016)
