
Abstract
Death receptors (DRs) are a growing family of transmembrane proteins that can detect the presence of specific extracellular death signals and rapidly trigger cellular destruction by apoptosis. Eight human DRs (Fas, TNF-R1, TRAMP, TRAIL-R1, TRAIL-R2, DR-6, EDA-R and NGF-R) have been identified. The best studied to date is Fas (CD95). Expression and signaling by Fas and its ligand (FasL, CD95L) is a tightly regulated process essential for key physiological functions in a variety of organs, including the maintenance of immune homeostasis. Recently, strong evidence has shown that dysregulation of Fas expression and/or signaling contributes to the pathogenesis of tissue destructive diseases such as graft-versus-host disease, toxic epidermal necrolysis, multiple sclerosis and stroke. With these new developments, strategies for modulating the function of Fas signaling have emerged and provided novel protein-based therapeutic possibilities that will be discussed herein. Selective triggering of DR-mediated apoptosis in cancer cells is an emerging approach that is being intensely investigated as a mode of cancer therapy. Local administration of Fas agonists, and more promisingly, systemic use of soluble recombinant forms of TRAIL have shown efficacy in preclinical models of the disease. Developments in this field that may have important clinical implications for the treatment of cancer are reviewed.
Similar content being viewed by others

Death receptors and their signaling
Cell death by apoptosis is a tightly regulated physiological process that enables the elimination of unwanted cells without causing an inflammatory response and its consequences. It is crucial for physiological processes such as embryonic development and the maintenance of tissue homeostasis, but when inappropriately regulated can contribute to tissue destructive disease and cancer for example.
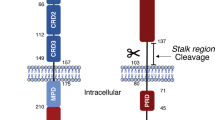
Certain cells are equipped with cell-surface sensors, named death receptors (DRs) that are able to detect the presence of specific extracellular death signals and frequently trigger cellular destruction by apoptosis. DRs are a subfamily of transmembrane proteins that belong to the tumor necrosis factor (TNF)-family of receptors. Eight human DRs (Fas (CD95), TNF-R1, TRAMP (WSL-1/Apo-3/DR-3/LARD), TNF-related apoptosis-inducing ligand (TRAIL)-R1 (DR-4), TRAIL-R2 (DR-5), DR-6, EDA-R and NGF-R) have been identified to date.1,2 All are type I membrane proteins that contain two to four cysteine-rich extracellular domains and a cytoplasmic motif named ‘death domain’ (DD) (Figure 1). The latter can couple the receptor to caspase cascades essential for the induction of apoptosis.

DRs and their ligands. Eight human DRs (Fas, TNF-R1, TRAMP, TRAIL-R1, TRAIL-R2, DR-6, EDA-R and NGF-R) are known to date. All are type I membrane proteins that contain two to four cysteine-rich extracellular domains and a cytoplasmic sequence named ‘death domain’ (DD). The known ligands for these DRs are shown. Some of the ligands undergo post-tranlational processing by metalloproteases or furin. DR6 is at present an orphan receptor
The best-studied DR signaling pathway to date is that triggered by the binding of Fas ligand (FasL) to Fas. Schematically, conformational changes within Fas occurring upon binding of the membrane-bound form of FasL recruits the bipartite adaptor molecule Fas-associated death-domain (FADD; composed of an amino terminal death effector domain (DED) and a carboxyl terminal DD). FADD binds to Fas (via homophilic DD–DD interactions) and recruits the DED-containing caspases-8 and -10 to the receptor via homophilic DED–DED interactions. Caspases-8 and -10 within this newly formed death-inducing signaling complex (DISC) then proteolytically autoactivate themselves and initiate apoptosis by subsequent cleavage of downstream effector caspases (caspases-3, -7) (Figure 2). Depending on the cell type and the intensity of the death signal, recruitment of adaptor molecules can be detected within minutes, and apoptosis completed within less than an hour. For Fas, there is an additional pathway that can signal cell death. It involves recruitment of RIP, a protein kinase that binds the DD of Fas and links this receptor to a cell death pathway that does not require activation of caspases.3 Although all DRs theoretically can transmit death signals, certain DRs including TNF-R1, TRAMP, DR6 and EDA-R preferentially transmit signals that induce specific gene expression, such as NF-κB activation. This can result in a diverse set of cellular responses including proliferation, differentiation and/or cellular activation, similarly to what classically occurs following signaling by TNF-R family members that do not have a DD (CD40, CD30, CD27…). For example, signaling of apoptosis through TNF-R1 usually occurs only if protein synthesis is blocked. This suggests that TNF triggering of apoptosis is under most circumstances actively suppressed by cellular inhibitory proteins such as FLIP or IAPs (Figure 2), thus privileging NF-κB or AP-1 mediated activation of proinflammatory and immunomodulatory genes.

The Fas signaling pathway and its molecular control. Fas signaling is triggered upon contact with membrane bound FasL. Subsequent recruitment of FADD and pro-caspase-8/10 leads to upstream caspase (caspase-8/-10) autoactivation which initiates apoptosis by subsequent cleavage of downstream effector caspases (caspases-3, -7). An alternative, caspase-independent pathway is initiated upon recruitment of the DD-containing kinase RIP1. DR apoptosis can be inhibited at the receptor level (antbodies, soluble ligands, etc.) by FLIPs (inhibiting caspase-8/-10) or by X-IAP (inhibiting caspases-3 (and -9)
DRs and disease: rationale for therapies targeting death receptors
To date, the implication of DRs in the pathogenesis of disease or in cellular defense mechanisms against disease has been best studied for the DRs to FasL and TRAIL. Herein we will focus on tissue destructive and cancerous diseases in which we consider that there is a window of opportunity for targeting Fas and/or TRAIL-mediated cell death with protein-based molecules as drug candidates.
Tissue destructive diseases and Fas-mediated cell death
Dysregulation of Fas signaling has been shown to contribute to tissue destruction in severe diseases such as graft-versus-host disease (GVHD), severe adverse drug reactions (toxic epidermal necrolysis), multiple sclerosis (MS), stroke, certain forms of hepatitis and large granular lymphocyte (LGL) leukemia. The evidence for Fas/FasL involvement in four of these diseases is summarized below.
Acute GVHD is a frequent complication of allogeneic bone marrow transplantation that results from the attack of transplanted donor lymphocytes against host organs including the skin, liver and gut. Although in its aggressive form, acute GVHD can be a serious life-threatening disease, in its mild form, the disease can be useful because of its antitumor effect. Increased apoptosis within target tissues is a key element of the pathogenesis of the immune-mediated skin, gut and liver damage in acute GVHD.4,5 Using cytolytic T lymphocytes (CTLs) from mice deficient in both functional FasL and perforin, it has been shown that the development of lethal acute GVHD in appropriate transplantation models can be virtually completely prevented.6,7 Delayed mortality was also observed in animals grafted with cells deficient in only one of the major cytolytic pathways (perforin or FasL), thus demonstrating that Fas-mediated cell death is implicated in the pathogenesis of acute GVHD.6
Toxic epidermal necrolysis (TEN, Lyell's syndrome) is a severe adverse drug reaction in which the abrupt onset of massive keratinocyte apoptosis results in the detachment of large sheets of epidermis from the underlying dermis and high patient mortality. It has recently been shown that this process is associated with highly increased keratinocyte FasL expression, together with conserved levels of keratinocyte Fas expression in vivo.8 Functional experiments performed by overlaying cryostat sections of lesional skin with Fas-sensitive cells as targets have demonstrated that keratinocyte FasL is cytolytically active in TEN. This cytolytic activity could be blocked with monoclonal antibodies that interfere with the interaction of Fas and FasL, thus supporting the hypothesis that increased keratinocyte FasL expression is responsible for the massive keratinocyte apoptosis that characterizes TEN. It also provided a rationale for molecular strategies to treat TEN.
MS is a progressive multifocal inflammatory demyelinating disease of the human central nervous system that affects over a million patients worldwide. Although the etiology of MS remains unclear, recent studies suggest that Fas signaling may contribute to the pathogenesis of MS by inducing selective oligodendrocyte and myelin destruction. First, increased Fas expression on T cells and an increase in soluble Fas in the cerebrospinal fluid of patients with MS have been reported.9,10 Second, Fas and FasL have been detected in MS lesions by immunohistochemistry,11 and lastly, in vitro data have demonstrated Fas expression on oligodendrocytes and induction of oligodendrocyte cell death by FasL.12 These results suggest that inhibitors of Fas may be useful in MS.
Stroke, a consequence of focal brain ischemia, affects 1.8 million people each year worldwide and is a common cause of death. It is known that during the first few hours after a stroke, neurons in the ischemic penumbra or peri-infarct zone, suffer transiently reversible damage and then ultimately undergo death by apoptosis.13 These first few hours of reversible neuron injury thus offer an opportunity for therapeutic intervention. DR signaling molecules FasL and TRAIL have been shown to be implicated in ischemia-induced neuronal apoptosis, and it has been suggested that Fas-mediated neuronal apoptosis in this situation is deleterious.14 Indeed, both lpr mice that lack functional Fas and gld mice that lack functional FasL are protected from ischemic neuronal injury. Deficiency in TNF appears to further improve the protection offered by deficient Fas signaling.14,15 The tissue destruction that occurs during stroke may therefore be to some extent limited by Fas inhibitors if used very early in the disease process.
Cancer, Fas and TRAIL
Defects in apoptosis contribute to both tumorigenesis and chemotherapeutic drug resistance. Tumorigenesis is a multistep process in which mutations in key cellular genes including those implicated in apoptosis produce a series of acquired advantages that allow the developing cancer cell to grow unchecked in the absence of growth-stimulating signals, while overcoming growth-inhibitory signals and the host immune response. The ability of cells to evade apoptosis is thus an essential feature of cancer development, progression and resistance to current therapeutic approaches.
Chemotherapy and radiation therapy are the conventional therapeutic tools used for the treatment of cancers that are not amenable to surgical cure. Although such treatments often result in tumor regression, they are rarely curative and are often hampered by the existence or emergence of resistant tumor cells. Mutations in p53 are frequently the cause of such resistance by preventing the signaling of apoptosis in response to cellular damage. Death ligands such as TRAIL and FasL, however, have been shown to be able to trigger apoptosis independently of p53 (for a review see Johnstone et al.,16).
Although the main role of TRAIL appears to be the induction of apoptosis, its range of physiological functions is probably still only incompletely understood. Several TRAIL receptors have been identified, notably two DRs TRAIL-R1 and TRAIL-R2) and three decoy receptors (TRAIL-R3 (DcR1), TRAIL-R4 (DcR2) and osteoprotegerin (OPG)) (for a review see Ashkenazi and Dixit1). TRAIL is constitutively expressed in many tissues. Furthermore, in peripheral blood T cells, NK cells and blood dendritic cells where it is not expressed in unstimulated state, TRAIL expression can be induced upon activation.2,17,18,19,20 Evidence suggests that TRAIL is implicated in activation-induced T-cell death, NK-mediated cytolysis and T-cell-mediated cytolysis of tumor cells. For example, mouse experiments in which TRAIL was blocked by neutralizing antibodies suggest that TRAIL is important for the antitumor activity of NK cells.21 Furthermore, recombinant soluble TRAIL has been shown to induce apoptosis in cell lines from a broad spectrum of human cancers, whereas most normal human nontumorigenic cell types are resistant to TRAIL with the exception of hepatocytes, neurons and astrocytes.22,23 Taken together it appears therefore that there is a strong scientific basis for developing therapeutic strategies that can mimic the activity of cell-bound TRAIL for use in cancer treatment.
Fas signaling is known to be implicated in CTL-mediated target cell death, and is thus a key element in the effector stage of an immune response against tumor cells that express Fas and have a functional Fas-signaling pathway. Fas expression at the cell surface is observed in cell lines derived from a variety of tumors, including glioma, colon carcinoma, breast carcinoma, prostate cancer, neuroblastoma, melanoma and medulloblastoma.24,25,26,27,28,29,30 Immunohistochemical studies have confirmed that such expression can be observed in tumors in vivo, or on freshly isolated tumor cells ex vivo. Functional assays in which such Fas expressing tumor cell lines are exposed to agonistic anti-Fas antibodies or soluble recombinant FasL have demonstrated that the Fas-signaling pathway is often functional and capable of transducing a death signal. Resistance to Fas signaling has been reported in tumor cells and includes Fas mutations which are usually quite rare, alternative splice variants of Fas, or the upregulation of apoptosis inhibitory proteins such as FLIP, DcR3, inhibitor of apoptosis proteins (IAPs) and Bcl-2 family members. Taken together, however, as abundant data showing sensitivity of certain tumor types to Fas exist, targeting Fas-mediated apoptosis by anti-Fas antibodies may be a promising anticancer therapy in vivo.
Cytotoxic drugs and tumor cell p53 status are known to have an influence on Fas-mediated apoptosis. In vitro, it has been demonstrated that chemotherapeutic drugs can induce Fas-mediated apoptosis and sensitize tumor cells to the cytolytic activity of FasL. Exposure of tumor cell lines to therapeutically relevant concentrations of cytotoxic drugs such as cisplatin, doxorubicin, etoposide, methotrexate or cytarabine upregulate tumor cell expression of Fas and FasL and Fas-mediated apoptosis in a manner that can be inhibited by antagonistic ant-Fas antibodies (for a review see Friesen et al. 31). In addition to cytotoxic drugs, cytokines such as interferons and TNF, can induce Fas expression and sensitize tumor cells to Fas-mediated apoptosis.
Functional wild-type p53 also promotes effective signaling of Fas-mediated apoptosis in many cell types (for a review see Johnstone et al.16). Several cytotoxic drugs are able to induce nuclear accumulation of wild-type p53 and simultaneously increase the surface expression of Fas and FasL in a variety of tumor cell types. Studies exist however suggesting that there is no link between p53 status and sensitivity to Fas-mediated apoptosis.
Potential for therapeutic inhibition of Fas signaling in disease
As reviewed above, DR pathways although only recently discovered, have already been implicated in the pathogenesis of several disease conditions. Developing strategies to inhibit or redirect these molecules for therapeutic purposes have therefore rapidly become a realistic and exciting challenge. Herein we will present a few examples of diseases in which such strategies hold significant potential for development and application in humans.
Tissue destruction in the skin, liver and gut correlates with clinical symptoms in GVHD, and is at least partially mediated by induction of Fas-mediated cell death by FasL positive donor lymphocytes that infiltrate these organs. In preclinical animal models of GVHD, tissue damage and the severity of GVHD can be inhibited by antibodies directed against FasL.32,33 Whether inhibition of acute GVHD by blockade of cytolytic pathways including Fas could be achieved without compromising the ‘beneficial’ graft versus leukemia (GVL) effect has only recently been assessed. Tsukada et al.34 have been able to demonstrate that selective inactivation of Fas, perforin or TNF all significantly inhibit murine acute GVHD, but only blockade of Fas does so in a manner that preserves the GVL reaction.35 Taken together, these preclinical mouse studies show that targeting of Fas or FasL with antagonistic antibodies has therapeutic potential in that it can selectively block GVHD without compromising GVL.
In TEN, the demonstration that epidermal keratinocyte FasL can induce Fas-mediated apoptosis in vitro that can be abrogated by blockade with monoclonal antibodies against FasL or by a hybrid protein consisting of Fas and the Fc portion of IgG (Fas: Fc), suggested that strategies aimed at inhibiting Fas signaling may be useful in the treatment of TEN. It has recently been shown that commercial preparations of intravenous immunoglobulins (IVIG) contain antibodies against Fas that are able to block the binding of FasL to Fas8 (Figure 3). Furthermore intravenous immunoglobulins, by blocking Fas, potently inhibit cell death mediated by recombinant FasL in vitro.8 When used in vivo at high doses (0.75 g/kg/day for 4 consecutive days) to treat patients with TEN, IVIG consistently and rapidly blocked the progression of skin detachment and disease,8 strongly suggesting that Fas blockade may be a viable therapeutic avenue for this life-threatening adverse drug reaction.

Schematic representation of the mechanism of inhibition of Fas-mediated cell death by IVIG. In normal skin (a), low levels of FasL are expressed by keratinocytes and localized intracellularly. In lesional skin during TEN (b), high levels of FasL are expressed by keratinocytes and localized at the cell surface. Upon contact with Fas, cell surface FasL induces Fas multimerization and rapid signaling of keratinocyte cell death by apoptosis. Inhibition of Fas-mediated keratinocyte apoptosis in the epidermis during TEN by IVIG because of the presence of naturally occurring antibodies that bind to and block the function of the Fas receptor (c)
The influence of Fas signaling on the course of experimental autoimmune encephalomyelitis (EAE), a model for MS, has been well investigated in mice. The frequency of EAE induced by myelin oligodendrocyte glycoprotein (MOG) is significantly lower in lpr mice lacking Fas than in wild-type mice (19 versus 94%). Furthermore, therapeutic blockade of Fas signaling by intrathecal administration of a neutralizing antibody against FasL during the progression phase of EAE has been shown to significantly reduce the severity of the disease by reducing inflammation and myelin destruction in the central nervous system.36 This data suggest that Fas or FasL inhibitors that are able to pass the hemato-encephalic barrier may be beneficial for suppression of acute inflammatory phases of MS in humans.
As Fas and TNF signaling appear to contribute to neuronal cell death after ischemic brain injury (stroke), infarct volumes and survival have been assessed in a mouse model of brain ischemia in the presence or absence of antibodies against FasL and TNF. Interestingly, mice injected with a mixture of neutralizing anti-TNF and anti-FasL antibodies (50 μg each) 30 min after induction of stroke showed a reduction in both infarct volumes and mortality by approximately 70%.15 Administration of either antibody alone did not significantly reduce infarct volumes or mortality in this setting however. This preclinical data suggest that therapeutic inhibition of Fas and TNF signaling may prove to be a viable therapeutic strategy in humans suffering from stroke.
Induction of DR-mediated apoptosis for cancer therapy
Two of the death ligands identified to date, FasL and TRAIL, have shown their ability to induce cancer cell death by apoptosis at least in vitro. Although the effect of FasL appears to be considerably less selective for cancer cells than TRAIL, we will briefly discuss the existing evidence for both as therapeutic candidates.
Triggering of Fas as a therapeutic approach for cancer
In 1989, Trauth et al. 37 demonstrated for the first time that triggering of Fas with an agonistic monoclonal antibody had potential for cancer therapy. In a human B-cell lymphoma transplanted to nude mice, intravenously administered anti-human Fas Ab rapidly induced tumor regression.37 Such an approach could not however be performed in mice with agonistic anti-mouse Fas Ab (Jo2) as all mice die within 6 h of injection because of the toxic effects resulting from massive engagement of Fas receptors in normal tissues including the liver.38 The systemic administration of Fas agonists such as agonistic anti-Fas Ab or recombinant FasL is at present impossible, without additional measures aimed at targeting these agents selectively to tumor, given the toxic side effects.
Local administration may hold some promise however. Rensing-Ehl et al.39 have shown that intraperitoneal injection of FasL-containing supernatant of FasL-transfected neuroblastoma cells could efficiently kill Fas-expressing murine lymphoma cells (Yac-1) implanted in capsules in the peritoneal cavity of mice without systemic toxicity.39 Thus, locally applied FasL can kill tumor cells very efficiently without systemic toxicity and may represent a candidate for local tumor treatment.
TRAIL-mediated apoptosis as an anticancer therapy
As described above, TRAIL is a potent and quite selective inducer of tumor cell apoptosis in vitro that mediates its signaling by binding to the DRs TRAIL-R1 and/or TRAIL-R2. In mouse models in vivo treatment with recombinant soluble TRAIL of immunodeficient mice bearing various human tumor types (including breast and colorectal carcinoma, myeloma, and glioma) has proven effective and safe.40,41,42,43 In addition, recent data suggest that recombinant TRAIL combined with cytotoxic drugs or radiation therapy can synergize to provide enhanced tumor destruction in mouse models.44,45
Current approaches for the development of TRAIL-R1 and/or TRAIL-R2 agonists as anticancer agents include antibody targeting of the receptor(s) and the use of soluble forms of TRAIL.
Recently, two groups have demonstrated that monoclonal Ab directed against either TRAIL-R1 or TRAIL-R2 can effectively inhibit tumor growth in vivo.46,47 Interestingly, in vivo studies performed with the anti-TRAIL-R2 antibody showed tumoricidal activity without hepatocyte toxicity thus confirming the selectivity of this therapeutic approach.
At least three distinct forms of soluble recombinant TRAIL with potential for therapeutic use have been reported to date. First, a Flag-tagged form of recombinant TRAIL generated by fusing the extracellular portion of TRAIL (residues 95–281) to an amino-terminal Flag-tag. This form of soluble recombinant TRAIL is rendered cytolytic for tumor cells when multimerized by crosslinking with an anti-Flag antibody.48 Although very active, this form of TRAIL has the disadvantage that it does not represent an ‘all in one’ solution for therapeutic application, since both recombinant Flag-tagged TRAIL and anti-Flag Ab are required to achieve appropriate signaling of apoptosis. A second form is LZ-TRAIL. This form of TRAIL was generated by fusing the extracellular portion of TRAIL (residues 95–281) to a modified yeast Gal-4 leucine zipper (LZ) that favors multimerization of TRAIL.40 This form or recombinant TRAIL provides an ‘all in one’ solution, and has been tested in preclinical animal models of cancer.40 Third, a fully human recombinant version of TRAIL consisting only of amino acids 114–281 of human TRAIL has been generated, and has recently shown promising results in preclinical and toxicology studies in small and large animals.49 Using this form of TRAIL, growth of established human colon carcinomas in mice injected intravenously on a daily basis with 30–120 mg/kg/day of TRAIL was significantly reduced (approx. seven-fold) as compared to controls. Pharmacokinetic studies, in mice, rats, cynomolgus monkeys, and chimpanzees revealed half-lives for i.v.-injected TRAIL ranging from 3 to 5 min in rodents and 23 to 31 min in nonhuman primates. No adverse effects were noted in cynomolgus monkeys and chimpanzees that recieved TRAIL, and predictions of the dose required for use in humans suggest that on a milligram per kilogram basis, doses significantly lower than those used in the above mouse xenograft studies could be effective in humans.
Conclusion and perspectives
Since the discovery of the first prototypic DR Fas in 1989, substantial progress has been made in our understanding of the mechanism of action and function of DRs. A total of eight DRs have now been identified and their downstream signaling pathways leading to cell death have been characterized, at least in part. Loss of function mutations in the Fas system have highlighted the importance of DR signaling in the maintenance of tissue homeostasis, specifically within the immune system. As outlined here, DRs such as Fas also contribute to disease. DRs are therefore double-edged swords. On the one hand, they protect tissues, and on the other, improper regulation of their function can result in rapid tissue destruction that can be life threatening, as illustrated by GVHD and TEN. Furthermore, as shown for TRAIL, selective use of DR signaling pathways can be a means of specifically targeting cancer cells.
Our current understanding of how DR-mediated apoptosis contributes to systemic disease has opened the path for designing protein-based drugs that target DR signaling pathways. Promising results have been already obtained to date with such drug candidates engineered to modulate Fas and TRAIL-receptor signaling pathways. Fas inhibitors hold great promise for the treatment of tissue destructive diseases such and GVHD, MS and stroke. TRAIL and FasL may prove to be useful for cancer therapy, either alone or in association with more conventional approaches such as chemotherapy and radiation therapy.
Abbreviations
- CTL:
-
cytotoxic T lymphocyte
- DcR:
-
decoy receptor
- DD:
-
death domain
- DED:
-
death effector domain
- DR:
-
death receptor
- EAE:
-
experimental autoimmune encephalitis
- FADD:
-
Fas-associated death domain protein
- FLICE:
-
FADD-like ICE (caspase-8)
- FLIP:
-
FLICE inhibitory protein
- GVHD:
-
graft-versus-host disease
- IAP:
-
inhibitor of apoptosis protein
- IVIG:
-
intravenous immunoglobulins
- MS:
-
multiple sclerosis
- RIP:
-
receptor-interacting protein
- TEN:
-
toxic epidermal necrolysis
- TRADD:
-
TNF receptor-associated death domain protein
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- TRAMP:
-
TNFR-related apoptosis-mediating protein
References
Ashkenazi A and Dixit VM . Apoptosis control by death and decoy receptors. Curr. Opin. Cell. Biol. 1999; 11: 255–260
Ashkenazi A and Dixit VM . Death receptors: signaling and modulation. Science 1998; 281: 1305–1308
Holler N, Zaru R, Micheau O, Thome M, Attinger A and Valitutti S et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000; 1: 489–495
Gilliam AC, Whitaker-Menezes D, Korngold R and Murphy GF . Apoptosis is the predominant form of epithelial target cell injury in acute experimental graft-versus-host disease. J. Invest. Dermatol. 1996; 107: 377–383
Sale GE . Does graft-versus-host disease attack epithelial stem cells. Mol. Med. Today 1996; 2: 114–119
Baker MB, Altman NH, Podack ER and Levy RB . The role of cell-mediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J. Exp. Med. 1996; 183: 2645–2656
Braun MY, Lowin B, French L, Acha-Orbea H and Tschopp J . Cytotoxic T cells deficient in both functional fas ligand and perforin show residual cytolytic activity yet lose their capacity to induce lethal acute graft-versus-host disease. J. Exp. Med. 1996; 183: 657–661
Viard I, Wehrli P, Bullani R, Schneider P, Holler N, Salomon D et al. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 1998; 282: 490–493
Ichikawa H, Ota K and Iwata M . Increased Fas antigen on T cells in multiple sclerosis. J. Neuroimmunol. 1996; 71: 125–129
Inoue A, Koh CS, Sakai T, Yamazaki M, Yanagisawa N, Usuku K et al. Detection of the soluble form of the Fas molecule in patients with multiple sclerosis and human T-lymphotropic virus type I-associated myelopathy. J. Neuroimmunol. 1997; 75: 141–146
Dowling P, Shang G, Raval S, Menonna J, Cook S and Husar W . Involvement of the CD95 (APO-1/Fas) receptor/ligand system in multiple sclerosis brain. J. Exp. Med. 1996; 184: 1513–1518
D'Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB et al. Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J. Exp. Med. 1996; 184: 2361–2370
Linnik MD, Zobrist RH and Hatfield MD . Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke 1993; 24: 2002–2008; discussion 2008–2009
Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J et al. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J. Neurosci. 1999; 19: 3809–3817
Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J et al. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001; 8: 679–686
Johnstone RW, Ruefli AA and Lowe SW . Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002; 108: 153–164
Fanger NA, Maliszewski CR, Schooley K and Griffith TS . Human dendritic cells mediate cellular apoptosis via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Exp. Med. 1999; 190: 1155–1164
Kayagaki N, Yamaguchi N, Nakayama M, Takeda K, Akiba H, Tsutsui H et al. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J. Immunol. 1999; 163: 1906–1913
Thomas WD and Hersey P . TNF-related apoptosis-inducing ligand (TRAIL) induces apoptosis in Fas ligand-resistant melanoma cells and mediates CD4 T cell killing of target cells. J. Immunol. 1998; 161: 2195–2200
Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES and Perussia B . Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998; 188: 2375–2380
Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Kayagaki N, Yagita H et al. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J. Exp. Med. 2002; 195: 161–169
Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR et al. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat. Med. 2000; 6: 564–567
Nitsch R, Bechmann I, Deisz RA, Haas D, Lehmann TN, Wendling U et al. Human brain-cell death induced by tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Lancet 2000; 356: 827–828
Weller M, Frei K, Groscurth P, Krammer PH, Yonekawa Y and Fontana A . Anti-Fas/APO-1 antibody-mediated apoptosis of cultured human glioma cells. Induction and modulation of sensitivity by cytokines. J. Clin. Invest. 1994; 94: 954–964
Tillman DM, Petak I and Houghton JA . A Fas-dependent component in 5-fluorouracil/leucovorin-induced cytotoxicity in colon carcinoma cells. Clin. Cancer Res. 1999; 5: 425–430
Ruiz-Ruiz MC and Lopez-Rivas A . p53-mediated up-regulation of CD95 is not involved in genotoxic drug-induced apoptosis of human breast tumor cells. Cell Death Differ. 1999; 6: 271–280
Rokhlin OW, Glover RA and Cohen MB . Fas-mediated apoptosis in human prostatic carcinoma cell lines occurs via activation of caspase-8 and caspase-7. Cancer Res. 1998; 58: 5870–5875
Terheyden P, Siedel C, Merkel A, Kampgen E, Brocker EB and Becker JC . Predominant expression of Fas (CD95) ligand in metastatic melanoma revealed by longitudinal analysis. J. Invest. Dermatol. 1999; 112: 899–902
Fulda S, Susin SA, Kroemer G and Debatin KM . Molecular ordering of apoptosis induced by anticancer drugs in neuroblastoma cells. Cancer Res. 1998; 58: 4453–4460
Fulda S, Scaffidi C, Pietsch T, Krammer PH, Peter ME and Debatin KM . Activation of the CD95 (APO-1/Fas) pathway in drug- and gamma-irradiation-induced apoptosis of brain tumor cells. Cell Death Differ. 1998; 5: 884–893
Friesen C, Fulda S and Debatin KM . Cytotoxic drugs and the CD95 pathway. Leukemia 1999; 13: 1854–1858
Hattori K, Hirano T, Miyajima H, Yamakawa N, Tateno M, Oshimi K et al. Differential effects of anti-Fas ligand and anti-tumor necrosis factor alpha antibodies on acute graft-versus-host disease pathologies. Blood 1998; 91: 4051–4055
Miwa K, Hashimoto H, Yatomi T, Nakamura N, Nagata S and Suda T . Therapeutic effect of an anti-Fas ligand mAb on lethal graft-versus- host disease. Int. Immunol. 1999; 11: 925–931
Tsukada N, Kobata T, Aizawa Y, Yagita H and Okumura K . Graft-versus-leukemia effect and graft-versus-host disease can be differentiated by cytotoxic mechanisms in a murine model of allogeneic bone marrow transplantation. Blood 1999; 93: 2738–2747
Schmaltz C, Alpdogan O, Horndasch KJ, Muriglan SJ, Kappel BJ, Teshima T et al. Differential use of Fas ligand and perforin cytotoxic pathways by donor T cells in graft-versus-host disease and graft-versus-leukemia effect. Blood 2001; 97: 2886–2895
Okuda Y, Sakoda S, Fujimura H, Nagata S, Yanagihara T and Bernard CC . Intrathecal administration of neutralizing antibody against Fas ligand suppresses the progression of experimental autoimmune encephalomyelitis. Biochem. Biophys. Res. Commun. 2000; 275: 164–168
Trauth BC, Klas C, Peters AM, Matzku S, Moller P, Falk W et al. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science 1989; 245: 301–305
Ogasawara J, Watanabe FR, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y et al. Lethal effect of the anti-Fas antibody in mice. Nature 1993; 364: 806–809
Rensing-Ehl A, Frei K, Flury R, Matiba B, Mariani SM, Weller M et al. Local Fas/APO-1 (CD95) ligand-mediated tumor cell killing in vivo. Eur. J. Immunol. 1995; 25: 2253–2258
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999; 5: 157–163
Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 1999; 104: 155–162
Mitsiades CS, Treon SP, Mitsiades N, Shima Y, Richardson P, Schlossman R et al. TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: therapeutic applications. Blood 2001; 98: 795–804
Pollack IF, Erff M and Ashkenazi A . Direct stimulation of apoptotic signaling by soluble Apo2l/tumor necrosis factor-related apoptosis-inducing ligand leads to selective killing of glioma cells. Clin. Cancer Res. 2001; 7: 1362–1369
Gliniak B and Le T . Tumor necrosis factor-related apoptosis-inducing ligand's antitumor activity in vivo is enhanced by the chemotherapeutic agent CPT-11. Cancer Res. 1999; 59: 6153–6158
Chinnaiyan AM, Prasad U, Shankar S, Hamstra DA, Shanaiah M, Chenevert TL et al. Combined effect of tumor necrosis factor-related apoptosis-inducing ligand and ionizing radiation in breast cancer therapy. Proc. Natl. Acad. Sci. USA 2000; 97: 1754–1759
Chuntharapai A, Dodge K, Grimmer K, Schroeder K, Marsters SA, Koeppen H et al. Isotype-dependent inhibition of tumor growth in vivo by monoclonal antibodies to death receptor 4. J. Immunol. 2001; 166: 4891–4898
Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat. Med. 2001; 7: 954–960
Bodmer JL, Meier P, Tschopp J and Schneider P . Cysteine 230 is essential for the structure and activity of the cytotoxic ligand TRAIL. J. Biol. Chem. 2000; 275: 20632–20637
Kelley SK, Harris LA, Xie D, Deforge L, Totpal K, Bussiere J et al. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J. Pharmacol. Exp. Ther. 2001; 299: 31–38
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
French, L., Tschopp, J. Protein-based therapeutic approaches targeting death receptors. Cell Death Differ 10, 117–123 (2003). https://doi.org/10.1038/sj.cdd.4401185
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401185
Keywords
This article is cited by
-
Semisynthetic aurones A14 protects against T-cell acute lymphoblastic leukemia via suppressing proliferation and inducing cell cycle arrest with apoptosis
Chinese Medicine (2022)
-
ITCH-dependent proteasomal degradation of c-FLIP induced by the anti-HER3 antibody 9F7-F11 promotes DR5/caspase 8-mediated apoptosis of tumor cells
Cell Communication and Signaling (2019)
-
USP8 suppresses death receptor-mediated apoptosis by enhancing FLIPL stability
Oncogene (2017)
-
Modulation of specificity protein 1 by mithramycin A as a novel therapeutic strategy for cervical cancer
Scientific Reports (2014)
-
A DR6/p75NTR complex is responsible for β-amyloid-induced cortical neuron death
Cell Death & Disease (2013)
