
Abstract
By crossing Huntington's disease (HD) R6/1 transgenic mice with ‘tissue’ transglutaminase (TG2) knock-out mice, we have demonstrated that this multifunctional enzyme plays an important role in the neuronal death characterising this disorder in vivo. In fact, a large reduction in cell death is observed in R6/1, TG2−/− compared with R6/1 transgenic mice. In addition, we have shown that the formation of neuronal intranuclear inclusions (NII) is potentiated in absence of the ‘tissue’ transglutaminase. These phenomena are paralleled by a significant improvement both in motor performances and survival of R6/1, TG2−/− versus R6/1 mice. Taken together these findings suggest an important role for tissue transglutaminase in the regulation of neuronal cell death occurring in Huntington's disease.
Similar content being viewed by others

Introduction
Transglutaminases catalyze Ca2+-dependent reactions in which the γ-carboxamide groups of peptide-bound glutamine residues serve as acyl donors and primary amino groups of several compounds function as acceptor substrates.1,2 The reaction results in post-translational modification of proteins by establishing ε(γ-glutamyl)lysine or N,N-bis(γ-glutamyl)polyamine crosslinks. TG-dependent formation of covalent crosslinking determines protein polymerisation, conferring resistance to mechanical breakage and chemical attack to the polymers.2
‘Tissue’ transglutaminase (TG2; E.C. 2.3.2.13) is one of the few genes specifically induced during physiological cell death.2 Its overexpression leads to a higher incidence of cell death in various mammalian cell lines, as TG2 transfected clones show a marked priming for induction of cell death,1,3,4 Conversely, transfection of various cell lines with an expression vector containing a segment of the human TG2 cDNA in antisense orientation resulted in a decrease in both spontaneous and induced death4,5.
It has also been demonstrated that TG2 may act as the Gαh subunit of the GTP-binding protein Gh, in a ternary complex with a 50 kDa subunit (Gαh), associated with the rat liver α1-adrenergic receptor.6 Thus, TG2/Gαh is a multifunctional protein, which by binding GTP in a GαhGTP complex, can shift its activity from cross-linking to receptor-mediated signalling, involving phospholipase-C (PLC) activation.6
Huntington's disease (HD) is a progressive autosomal dominant neurodegenerative disorder, resulting from the expansion of a trinucleotide (CAG) repeats (beyond 35–40 residues) in the coding region of huntingtin (htt).7 This event results in an exceedingly high number of contiguous glutamine residues in the translated protein, which gains a new function toxic for neurons. The disease is characterised by the formation of misfolded ubiquitinated protein aggregates (neuronal intranuclear inclusions, NII), containing all or part of the mutant htt.8 These protein inclusions have predominantly been detected in the nucleus, but may also be observed in the cytoplasm of striatal and cortical neurons.8 Several reports suggest that in vitro aggregation may occur both through polar zippers formation and/or protein cross-linking catalysed by TG2.9,10 Recent studies showed that TG2 does not interact with mutant huntingtin in cellular models and it is not involved in the assembly of NIIs,11 but despite these in vitro indications, no in vivo evidence for either models was so far obtained.
The aim of this study was to investigate the role of TG2 in vivo, both in the formation of NIIs and in the onset of the neuronal death at the basis of Huntington's pathogenesis.
Results and Discussion
TG2 is the predominant TGase in mouse brain
In order to elucidate the role of TG2 in Huntington's disease, we generated a novel animal model by crossing the R6/1 strain transgenic for exon 1 of huntingtin, carrying 116 (CAG) repeats, the disease onset of which is observed at about 16 weeks (Figure 1a,b),12 with a TG2 knock-out strain13 (Figure 1a,c,d). Initially, we studied the effect of the TG2 ablation on the weight loss characterizing HD pathogenesis (Figure 1e). The deletion of TG2 led to significant amelioration in weight loss which is particularly evident at 25 weeks of age (P<0.01 ANOVA test; P<0.05 post hoc test). A tendency to reduced weight loss was also detected in the brains of R6/1,TG2−/− mice when compared with those of R6/1 transgenic animals (Figure 1f). No statistically significant differences were observed both in total body and brain weight in the R6/1 when compared with R6/1, TG2+/− mice (Figure 1e,f).

Characterisation of HD R6/1,TG−/− mice. (a) Genomic DNA PCR analyses: presence of htt exon 1 transgene12 and TG2 gene disruption.13 Lane 1: R6/1. Lane 2: wild-type Lane 3: R6/1, TG2−/−. Lane 4: TG2−/−. Lane 5: R6/1, TG2−/−. Lane 6, molecular markers (marker V, ROCHE). (b) Western blot analysis showing the expression of htt exon 1 in full brain homogenates (n=5). No major differences were detectable between R6/1 and R6/1, TG2−/− mice. Lane 1: R6/1. Lane 2: R6/1, TG2−/−. Lane 3: TG2−/−. Lane 4: wild-type. Lower panel shows β-actin levels. (c) Western blot analysis showing the expression of TG2 in total brain homogenates (n=4). Lane 1: wild-type; Lane 2: R6/1; Lane 3: TG2+/−; Lane 4: R6/1, TG2+/−; Lane 5: TG2 −/−; Lane 6: R6/1, TG2−/−. (d), Western blot analysis showing the expression of TG2 in neocortex and striatum brain homogenates (n=5). Lane 1: wild-type striatum; Lane 2: wild-type cortex; Lane 3: R6/1 striatum; Lane 4: R6/1 cortex; Lane 5: TG2−/− striatum; Lane 6: TG2−/− cortex; Lane 7: R6/1, TG2−/− striatum; Lane 8: R6/1, TG2−/− cortex. (e), Histogram showing the mice body weights. R6/1, TG2−/− mice show a less severe loss of weight when compared to R6/1 animals (n=15; 30 weeks-old) ANOVA showed significant effect of genotype (F (5/29): 1145 733, P<0.01). Post-hoc comparisons (Tukey HSD) indicated a significant effect of genotype(P<0.05). (f) Histogram showing brain weights. A less pronounced loss of weight was detectable in R6/1, TG2−/− mice when compared to R6/1 ones (n=15; 30 week-old). ANOVA showed significant effect of genotype (F (5/29): 66 910, P<0.01). Post-hoc comparisons (Tukey HSD) indicated a significant effect of genotype (P<0.05)
In order to study the relative contribution of TG2 activity with the other TGases in mouse brain, we compared the TGase activity in brains of wild-type, TG2−/−, R6/1 and R6/1,TG2−/− mice both in the presence and absence of GTP, which specifically inhibits TG2 activity. Data reported in Figure 2a indicate that TG2 is the predominant TGase active in the brain. In fact, its ablation leads to an almost complete inhibition of activity in vitro detected as GTP-dependent putrescine incorporation into casein (Figure 2a). In order to verify whether this effect could also be observed in vivo, we compared the levels of the ε(γ-glutamyl)lysine cross-links in the insoluble fraction of TG2−/− mice brains with those detected in wild-type and in R6/1 transgenic animals (Figure 2b).14 By using this approach we detected that the htt transgene expression induces a marked increase in the ε(γ-glutamyl)lysine bond levels compared to controls, thus indicating that the accumulation of polyglutamine tracts stimulate the TG2 cross-linking activity in vivo. It is important to note that this increase is not detectable by measuring TGase activity in vitro (compare Figure 2a versus b) since this increase in the levels of cross-links is due to the activation of the enzyme activity in situ and not to increased TG2 protein levels as shown in Figure 1c,d. In keeping with these findings, the depletion of TG2 in HD transgenic mice results in a drastic reduction in ε(γ-glutamyl)lysine bond levels (Figure 2b). The drastic decrease (10-fold) in the isodipeptide levels observed in the R6/1, TG2−/− mouse brain (Figure 2b), also evidenced by the profound modification of the TG substrates pattern in mouse brain (Figure 2c), provides the first in vivo evidence of the predominant role played by TG2 among the various transglutaminases in mouse brain, as well as the absence of compensation in TG2−/− mice.

In vivo and in vitro detection of TGase activity in wild-type, R6/1,TG+/+ and R6/1,TG−/− mice. (a) TGases activity detection in vitro. The presence of the transgene does not significantly increase the levels of activity measured by supplying exogenous substrates, whereas in TG2−/− and R6/1,TG2−/− genotypes the expected drastic reduction was observed. When 0.5 mM GTP was added, no activity was detected. Each column represents the mean±S.D. of triplicate measurements in duplicate of total brain homogenates obtained from 25 week-old animals. (b) HPLC analysis of ε(γ-glutamyl)lysine bonds levels in the detergent-insoluble fraction of 25-week-old mouse brains (n=3). Data are statistically significant (P<0.005, Student's t-test) when compared to wild-type mice. (c) Western blot analysis of total TG substrate proteins. Aliquots of 25-week-old mouse brain homogenates were incubated with a TG biotinylated pansubstrate (5-(biotinamido)pentylamine, EZ-Link, Pierce) at 37°C for 1 h and biotinyl-substrates were revealed by HRP-streptavidin (Amersham)
TG2 ablation might, indeed, lead to an accumulation of its substrates. In absence of TG2, the number of proteins which might act as acyl donors in the transamidating reaction drastically decreases (Figure 2c), thus demonstrating a strict TG2 specificity for some transglutaminases substrates.
Finally, several evidences show that mutated huntingtin is a TG2 substrate in vitro.15 Since mutant htt is a predominant component of NII, the significant increase of aggregates in absence of TG2 (Figure 3c) might well reflect a substrate accumulation (Figure 2c).

TG2 ablation leads to an increased number of NIIs in vivo. (a) Epon embedded, toluidine blue-stained semithin section from 25-week old R6/1 mouse showing neurons in the primary motor cortex. Aggregates appear as white spots in the neuronal nuclei (arrows) (500×). (b) Ubiquitin immunohistochemistry showing red stained NIIs in R6/1,TG2−/− 25 week-old mouse striatum. (c) Percentage of NIIs detected in the striatum of R6/1, R6/1,TG2+/− and R6/1,TG2−/− mice (n=3). It is important to note that TG2−/− mice do not show any NIIs in the brain regions. Immunohistochemistry was performed at different ages, using an anti-ubiquitin antibody (clone FPM1, Ylem) (200×). ANOVA showed significant effect of genotype (F (2/12): 66.91, P<0.05). Post-hoc comparisons (Tukey HSD) indicated a significant effect of genotype (P<0.05)
Consistent with these data, it has recently been shown that ε(γ-glutamyl)lysine isodipeptide levels significantly increase in the cerebrospinal fluid of HD patients.16
TG2 ablation leads to an increased number of NIIs in vivo
To verify the TG2 involvement in NII formation in vivo, we evaluated the occurrence of NIIs in R6/1 versus R6/1,TG2−/− neocortex and striatum. The inclusions were revealed both on toluidine blue-stained semithin sections (Figure 3a) and on paraffin sections processed for ubiquitin immunolocalization (Figure 3b), in order to be sure that no difference in NII ubiquitination occurred in the absence of TG2. Interestingly, ablation of TG2 does not prevent NII assembly (Figure 3c); by contrast, we observed a statistically significant increase (30%) in the NII in R6/1 versus R6/1, TG2−/− brains (Figure 3c; TG2−/− mice do not show any NII in the brain, data not shown). Indeed, it is surprising that such increase is associated to clinical amelioration. Nevertheless, several evidences show that NII formation is not correlated with cell death and, more over, it has been hypothesised a protective role for aggregates.17,18 Our results provide an in vivo evidence in the direction of these findings, which also indicate that TG2 crosslinking of the mutated htt is not directly involved in NII assembly in vivo and suggest a complex role for TG2 in the regulation of the assembly of large nuclear protein complexes. The regulation of this phenomenon might also be determined by the inactivation of the TG2/Gαh receptor-mediated signalling pathway and/or TG2 integrin-binding adhesion functions.6,19
TG2 ablation reduces cell death in HD R6/1 transgenic mice
TG2 gene induction characterises cells undergoing cell death in either physiological and experimental settings.1,3,4 In keeping with these findings, we investigated whether TG2 ablation affects neuronal death levels in R6/1 versus R6/1, TG2−/− mice. In previous studies, both other groups20 and our own,21 have described the cell death in the cortex and striatum of R6/1 transgenic mice as being characterised by the appearance of condensed neurons not displaying the classical apoptotic features (Figure 4a). In order to gain further insight on the type of cell death involved in HD pathogenesis as well as to verify whether TG2 ablation could cause alteration in the cell death type, we ultrastructurally characterised neuronal death, in cortical and striatal brain regions, in animals expressing the mutated htt transgene both in the presence and absence of TG2. Independently of the age (18 and 25 weeks) or the genotype (R6/1 or R6/1, TG2−/−) of the animals analysed, the cytoplasm of suffering neurons showed numerous lysosomes and dilated endoplasmic reticulum (Figure 4b). This finding, which was not detected in normal mouse brain, suggests the involvement of autophagy in HD. In keeping with this hypothesis, mutated htt has been demonstrated to stimulate autophagy in cellular models.22 To investigate whether TG2 is involved in the formation of these ‘abnormal’ cells in vivo, we evaluated their levels in R6/1 versus R6/1, TG2−/− neocortex and striatum (Figure 4c). Results showed a dramatic reduction (60–70%) in the number of these condensed suffering cells in both brain regions of R6/1, TG2−/−, as compared to htt transgenic animals (Figure 4c). In a subsequent set of experiments we monitored the expression of calbindin D-28K (Figure 4d,e), a biochemical marker of neuronal viability,23 both on total brain (Figure 4d), as well as in neocortex and striatum (Figure 4e). Noteworthy, we detected that the reduction in the calbindin D-28K protein levels observed in the R6/1 mouse strain, indicating a neuronal cell loss, was much less pronounced in R6/1, TG2−/− mouse brains. These data indicate that the ablation of TG2 ameliorates the neurodegenerative process observed in HD mice and suggest that the TG2-dependent changes observed in the condensed suffering neurons are associated with neurodegeneration.

TG2 is an effector element of neuronal cell death in vivo. (a) Progressive condensation of striatal neurons as detected in semithin sections obtained from R6/1 mice. London Resin White (LWR) embedded, toluidine blue-stained semithin section. Healthy neuron, displaying a clear cytoplasm and a euchromatic nucleus.1 Abnormal neurons at different stages of condensation2,3,4 (700×). (b) Electron micrographs of a condensed neuron from a 25 week-old R6/1 mouse striatum, showing dilated smooth ER membranes (double-arrowhead) in the cytoplasm; an intranuclear inclusion is visible (asterisk) near a nucleolus (nu) (12 000×). (c) Percentage of ‘abnormal’ cells in R6/1, R6/1,TG2+/− and R6/1,TG2−/− mice (n=4), at various ages. A total number of 20 600 cells were examined. Data are statistically significant (P<0.01, Student's t-test) when compared to their respective controls (R6/1). (d) Western blot analysis of calbindin D-28k levels in 25-week-old mice (n=3) was carried out on 25 μg of total brain extracts by using CL-300 monoclonal antibody (SIGMA). Lower panel, loading controls revealed by β-actin polyclonal antibody (A2066, SIGMA). The histogram shows the intensity of calbindin D-28k signal when normalised with that derived from β-actin (n=4, P<0,01, Student's t-test). From left to right: Lane 1: wild-type; Lane 2: R6/1; Lane 3: TG2−/−; Lane 4: R6/1, TG2−/−. (e) Western blot analysis of calbindin D-28k levels in 25-week-old mice (n=4) was carried out on 25 μg of neocortex and striatum extracts by using CL-300 monoclonal antibody (SIGMA). Lower panel, loading controls revealed by β-actin polyclonal antibody (A2066, SIGMA). From left to right: Lane 1: wild-type; Lane 2: R6/1; Lane 3: TG2−/−; Lane 4: R6/1, TG2. From left to right: Lane 1: wild-type striatum; Lane 2: wild-type cortex; Lane 3: R6/1 striatum; Lane 4: R6/1 cortex; Lane 5: TG2−/− striatum; Lane 6: TG2−/− cortex; Lane 7: R6/1, TG2−/− striatum; Lane 8: R6/1, TG2−/− cortex. (f) Life span was analyzed in the wild-type, R6/1 and R6/1,TG2−/− animals (n=7) of the same age. The animals were maintained in the same conditions of lightning, temperature and feeding
TG2 ablation prolongs survival and improves motor performances in HD R6/1 mice
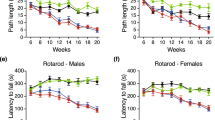
It is relevant to note that the significant reduction in neurodegeneration observed in R6/1, TG−/− mice leads to major differences both in the symptoms and survival of these animals. In fact, while all the R6/1 died after 360±8 days, R6/1, TG2−/− died at 416±8 days of age, with a significant increase of about 12% of their life span (P<0.01 Student's t-test) (Figure 4f). It is well known that Huntington's disease symptoms are characterised by chorea and thus alteration of motor functions, which can be monitored in animal models by the rotarod test.24 A significant amelioration in the motor behaviour of the R6/1,TG2−/− mice paralleled the improvement in the overall survival, thus indicating that TG2 is a major biochemical effector element of the cell death programme leading to HD-associated neurodegeneration. In fact R6/1,TG2−/− mice were able to stay on the rota rod for a significantly longer time period in comparison with R6/1 animals (at week-30: R6/1=3.5 s R6/1,TG2−/−=9.8 s). An improvement in motor behaviour, weight loss and life span have been very recently reported in a similar HD transgenic mouse model (R6/2) by using a putative pan-TGases inhibitor cystamine.25
Taken together our findings indicate that TG2 is the most relevant protein crosslinking enzyme among the various members of the transglutaminase family expressed in mouse brains. In addition, we showed, for the first time in vivo, that the TG2 plays a central complex role in the neurodegeneration process characterising HD pathogenesis. It is very likely that TG2 acts quite early in course of HD pathogenesis, as indicated by the reduced number of ‘abnormal’ cells detected at 18 weeks of age in HD mice lacking TG2. Furthermore, the fact that an increased level of NIIs is associated with a general improvement of the Huntington's disease transgenic mice favours the hypothesis that aggregate formation acts as protective event preventing and/or delaying neuronal cell death onset. Finally, the amelioration of both symptoms and survival, observed in R6/1, TG2−/− mice together with the notion that identical effects are obtained by treating a different HD mouse model (R6/2) with cystamine,25 indicates that the inhibition of this enzyme may be a novel approach for therapy of diseases caused by accumulation of polyglutamine repeats, including Huntington's disease.
Materials and Methods
Animals
All animals were housed and handled according to guidelines proposed by the Society of Neuroscience and the Italian National Research Council. Heterozygous R6/1 mice of the CBAxC57BL/6 strain were purchased by the Jackson laboratories and maintained by crossing carrier males with C57BL/6 females (eight backcross when mated with TG mice). Heterozygous TG2+/− mice of the SWJ129xC57BL/6 strain were back-crossed five times with C57BL/6 mates. TG2+/− mice were then subsequently crossed to generate knock-out individuals. To generate the R6/1, TG2 colony, carrier R6/1 males were mated with TG2−/− females. R6/1, TG2+/− mice were then crossed with TG2+/− animals to generate both R6/1,TG2−/−, R6/1,TG2+/− and R6/1,TG2+/+ littermates. Of the three lines generated by different founders no major differences were detected. Before experiments mice were deeply anaesthetised with tribromoethanol (Avertin; 0.2 ml of 1.25% solution/10 g of body weight) before being rapidly killed by transcardial perfusion with the fixative solution in order to prepare the samples for microscopy, or killed by cervical dislocation for biochemical analysis.
Transglutaminase activity assay
Brains from the various mice analyzed (25 weeks old), were washed extensively with phosphate buffered saline (PBS without Ca2+ and Mg2+) and homogenised in 0.1 M Tris-HCl, pH 7.5, containing 0.25 M sucrose, 0.5 mM EDTA, 1 mM PMSF. TGase activity was measured by detecting the incorporation of (3H)putrescine into N,N′-dimethylcasein as previously described.4 Protein concentration was determined using bovine serum albumin as standard.4
Western blot analysis
Mice were deeply anaesthetised and sacrificed. Brains were extracted immediately and dissected under a Zeiss ZSO stereomicroscope. Striatum and full cortex were isolated. Tissues were homogenised in sucrose 250 mM, Tris HCl pH 7.4 20 mM and EDTA 1 mM and protease inhibitors (Complete, EDTA free, ROCHE). Fifty μg of crude homogenate were separated on 12% SDS-polyacrilammide gel and transferred to nitrocellulose membrane. Membranes were probed with monoclonal mouse antibody specific for TG2 (AB3, CUB7402+TG100, Neomarkers-1 : 1000), monoclonal mouse antibody specific for calbindinD-28k (CL-300, SIGMA-1 : 1500), monoclonal mouse antibody specific for polyglutamine tracts (1C2, Euromedex-1 : 25 000) and polyclonal specific for β-actin (A2066, SIGMA-1 : 2000). The signals were detected with Supersignal (PIERCE) and the intensity of the bands measured and normalised with the NIH Image 1.62 programme.
Western blot analysis of total TGases substrate proteins
Aliquots of 25-week-old mice brain homogenates (10 mg) were incubated with a TG biotinylated pansubstrate (5-(biotinamido)pentylamine, EZ-Link, Pierce) at 37°C for 1 h (Tris-HCl pH 8.3 150 mM, CaCl2 15 mM, NaCl 90 mM, DTT 30 mM, EZ-Link 30 mM). Control reaction was performed in presence of EDTA 30 mM. Biotinyl-substrates (100 μg) were purified and analyzed by Western blot analysis, revealed by HRP-streptavidin (Amersham-1 : 1000).
Analysis of ε(γ-glutamyl)lysine isodipeptide levels
Fresh brain homogenates were centrifuged at 800 g, 4°C for 15 min. The pellet was resuspended in sucrose 0.33 M, Tris-HCl 0.2 M pH 7.5 and centrifuged at 1000 g, 4°C for 15 min. Subsequently, the pellet was resuspended in Triton X-100 1%, deoxycholic acid (DOC) 0.25%, guanidine 8 M and centrifuged at 4000 g, 4°C, for 15 min. Following three washes with distilled water the levels of ε(γ-glutamyl)lysine bonds levels in each sample were evaluated by HPLC as previously described.14
Light microscopy and immunohistochemistry
Mice were perfused at room temperature with phosphate-buffered saline pH 7.3, followed by 4% freshly depolymerised paraformaldehyde in 0.1 M phosphate buffer, pH 7.3. The brains were removed 1 h after perfusion. Brains were dehydrated and paraffin embedded following standard procedure and saggital sections cut on a microtome (Leitz) at 6 μm. Immunohistochemical staining was performed using monoclonal mouse antibody specific for ubiquitin (clone FPM1, YLEM). Incubations with the primary antibody were carried out in a wet chamber overnight at 4°C. A biotinylated goat anti-mouse IgG was used as secondary antibody, followed by a preformed strept-avidin-horseradish peroxidase complex (BioGenex, USA). The reaction was developed using aminoethylcarbazole (AEC), (CRL, USA) as chromogen substrate and 0.01% H2O2. Endogenous peroxidase activity was blocked by methanol-H2O2.
Electron microscopy
After perfusion, saggital brain sections were cut on a Vibratome at 40 μm and collected in phosphate-buffered saline pH 7.3. Slices were post-fixed in 1% OsO4 in 0.1 M phosphate buffer, rinsed in water, dehydrated, and flat embedded in Epon. Selected areas were then remounted on Epon blanks and sectioned on a Reichert Ultracut S ultramicrotome. Semithin sections were stained with tolouidine blue and photographed using a Axioplan 2 Zeiss light microscope. Ultrathin sections were briefly contrasted with uranyl acetate and lead citrate and photographed in a Philips CM120 electron microscope. For LR White embedding, corpus striatum and hippocampus were isolated from brains and cut in smaller pieces. Samples were rinsed in buffer, partially dehydrated (up to 95% alcohol), and embedded in London Resin White (LR White, Agar Scientific Ltd., Stanted, UK). Tissue blocks were cut on a Reichert Ultracut S ultramicrotome, ultra-thin sections were contrasted with uranyl acetate and lead citrate and photographed in a Philips CM120 electron microscope.
Statistical analysis
Cell counting was performed on blind-coded slides by three independent individuals. data are presented as mean±S.D. The statistical significance of the body weight, brain weight, NII number and latency to fall data were carried out by ANOVA and post hoc test (Turkey HSD). All other data significance was assessed with Student's t-test.
Blue tolouidine stained semithin sections from corpus striatum and hippocampus of R6/1, R6/1, TG2+/−, R6/1, TG2−/− and wild type mice (n=6) were examined using a Axioplan 2 Zeiss light microscope, at low-to-high magnifications. Different embedding blocks were analyzed, and for each block a minimum of 100 cells were observed. The percentage of ‘abnormal’ cells versus the total cell number was evaluated considering as altered those neurons showing high condensation and strong nuclear basophylia. Cells showing mild shrinkage and nuclear staining were classified separately. A total of 20 600 cells were counted.
The percentage of cells containing intranuclear inclusions in the striatum of R6/1, R6/1,TG2+/−, R6/1,TG2−/− animals (n=6) was evaluated on immunohistochemical sections, using a set of three slides for each mouse and a Zeiss Axioskop 20 connected to a JVC video system with a counting frame on the monitor. For each slide, 10 fields within the striatum, were analyzed. A total number of 10 400 cells were counted.
Intranuclear inclusions were also counted with the same criteria on blue tolouidine stained semithin sections using a Axioplan 2 Zeiss light microscope, at low-to-high magnifications. Since NII are similar in size with nucleoli and each cell contains two nucleoli, the number of cells showing intranuclear inclusion was compared with that derived from cells showing nucleolus.
Abbreviations
- HD:
-
Huntington's disease
- NII:
-
neuronal intranuclear inclusions
- TG2:
-
type 2 transglutaminase
- htt:
-
huntingtin
- ANOVA:
-
analysis of variance between groups
References
Melino G, Piacentini M . 1998 ‘Tissue’ transglutaminase in cell death: a downstream or a multifunctional upstream effector? FEBS Lett. 430: 59–63
Piacentini M, Colizzi V . 1999 Tissue transglutaminase: apoptosis versus autoimmunity Immunol. Today 20: 130–134
Gentile V, Thomazy V, Piacentini M, Fesus L, Davies PJ . 1992 Expression of tissue transglutaminase in Balb-C 3T3 fibroblasts: effects on cellular morphology and adhesion J. Cell Biol. 119: 463–474
Oliverio S, Amendola A, Rodolfo C, Spinedi A, Piacentini M . 1999 Inhibition of ‘tissue’ transglutaminase increases cell survival by preventing apoptosis J. Biol. Chem. 274: 34123–34128
Melino G, Annicchiarico-Petruzzelli M, Piredda L, Candi E, Gentile V, Davies PJ, Piacentini M . 1994 Tissue transglutaminase and apoptosis: sense and antisense transfection studies with human neuroblastoma cells Mol. Cell. Biol. 14: 6584–6596
Nakaoka H, Perez DM, Baek KJ, Das T, Husain A, Misono K, Im MJ, Graham RM . 1994 Gh: a GTP-binding protein with transglutaminase activity and receptor signaling function Science 264: 1593–1596
The Huntington's Disease Collaborative Research Group. 1993 A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes Cell 72: 971–983
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N . 1997 Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain Science 277: 1990–1993
Perutz MF, Johnson T, Suzuki M, Finch JT . 1994 Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases Proc. Natl. Acad. Sci. USA 91: 5355–5358
Kahlem P, Green H, Djian P . 1998 Transglutaminase action imitates Huntington's disease: selective polymerization of Huntingtin containing expanded polyglutamine Mol. Cell 1: 595–601
Chun W, Lesort M, Tucholski J, Ross CA, Johnson GV . 2001 Tissue transglutaminase does not contribute to the formation of mutant huntingtin aggregates J. Cell Biol. 153: 25–34
Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper B, Hetherington C, Lawton M, Trottier Y, Lehrach H Davies SW, Bates GP . 1996 Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice Cell 87: 493–506
De Laurenzi V, Melino G . 2001 Gene disruption of tissue transglutaminase Mol. Cell. Biol. 21: 148–155
Tarcsa E, Fesus L . 1990 Determination of epsilon (gamma-glutamyl)lysine crosslink in proteins using phenylisothiocyanate derivatization and high-pressure liquid chromatographic separation Anal. Biochem. 186: 135–140
Karpuj MV, Garren H, Slunt H, Price DL, Gusella J, Becher MW, Steinman L . 1999 Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington's disease brain nuclei Proc. Natl. Acad. Sci. USA 96: 7388–7393
Jeitner TM, Bogdanov MB, Matson WR, Daikhin Y, Yudkoff M, Folk J E, Steinman L, Browne SE, Beal M F, Blass J P, Cooper A J . 2001 N(epsilon)-(gamma-L-glutamyl)-L-lysine (GGEL) is increased in cerebrospinal fluid of patients with Huntington's disease J. Neurochem. 79: 1109–1112
Saudou F, Finkbeiner S, Devys D, Greenberg ME . 1998 Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions Cell 95: 55–66
Simeoni S, Mancini MA, Stenoien DL, Marcelli M, Weigel NL, Zanisi M, Martini L, Poletti A . 2000 Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract Hum. Mol. Genet. 9: 133–144
Akimov SS, Krylov D, Fleischman LF, Belkin AM . 2000 Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin J. Cell Biol. 148: 825–838
Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW . 2000 Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease Proc. Natl. Acad. Sci. USA 97: 8093–8097
Iannicola C, Moreno S, Oliverio S, Nardacci R, Ciofi-Luzzatto A, Piacentini M . 2000 Early alterations in gene expression and cell morphology in a mouse model of Huntington's disease J. Neurochem. 75: 830–839
Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M . 2000 Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy J. Neurosci. 20: 7268–7278
Kiyama H, Seto-Ohshima A, Emson PC . 1990 Calbindin D28K as a marker for the degeneration of the striatonigral pathway in Huntington's disease Brain Res. 525: 209–214
Luesse HG, Schiefer J, Spruenken A, Puls C, Block F, Kosinski CM . 2001 Evaluation of R6/2 HD transgenic mice for therapeutic studies in Huntington's disease: behavioral testing and impact of diabetes mellitus Behav. Brain Res. 126: 185–195
Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, Mitchell D, Steinman L . 2002 Prolonged survival and decreased abnormal movements in transgeni model of Huntington disease, with administration of the transglutaminase inhibitor cystamine Nat. Med. 8: 143–149
Acknowledgements
The authors would like to express their gratitude to Professors F Autuori and A Ciofi-Luzzatto for stimulating discussions, Dr. Enrico Girardi for the statistical analysis, Valentina Imbroglini for technical assistance and Dr. G Citro from Istituto Regina Elena, Rome, Italy for animal care and stabulation. The work was partially supported by grants from: European Community ‘Apoptosis Mechanisms’, AIRC. The financial support of Telethon-Italy (Grant n. E.1257 to F Bernassola) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by R A Knight
Rights and permissions
About this article
Cite this article
Mastroberardino, P., Iannicola, C., Nardacci, R. et al. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ 9, 873–880 (2002). https://doi.org/10.1038/sj.cdd.4401093
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401093
Keywords
This article is cited by
-
Probing tissue transglutaminase mediated vascular smooth muscle cell aging using a novel transamidation-deficient Tgm2-C277S mouse model
Cell Death Discovery (2021)
-
Discovery of sultam-containing small-molecule disruptors of the huntingtin–calmodulin protein–protein interaction
Medicinal Chemistry Research (2020)
-
Structural aspects of transglutaminase 2: functional, structural, and regulatory diversity
Apoptosis (2017)
-
Role of tissue transglutaminase-2 (TG2)-mediated aminylation in biological processes
Amino Acids (2017)
-
Transglutaminase 2 has opposing roles in the regulation of cellular functions as well as cell growth and death
Cell Death & Disease (2016)
