
Abstract
The induction of apoptosis in human keratinocytes by UV radiation involves caspase-mediated cleavage and activation of protein kinase C delta (PKCδ). Here we examined the role of PKC activation in caspase activation and disruption of mitochondria function by UV radiation. Inhibition of PKC partially blocked UV radiation-induced cleavage of PKCδ, pro-caspase-3, and pro-caspase-8, and the activation of these caspases. PKC inhibition also blocked the UV-induced loss of mitochondria membrane potential, but did not block the release of cytochrome c from mitochondria. Expression of the active catalytic domain of PKCδ was sufficient to induce apoptosis and disrupt mitochondrial membrane potential, however a kinase inactive PKCδ catalytic domain did not. Furthermore, the PKCδ catalytic fragment generated following UV radiation localized to the mitochondria fraction, as did ectopically expressed PKCδ catalytic domain. These results identify a functional role for PKC activation in potentiating caspase activation and disrupting mitochondrial function during UV-induced apoptosis.
Similar content being viewed by others

Introduction
Induction of programmed cell death, or apoptosis, by UV radiation is a major cellular protective mechanism for skin from the carcinogenic effects of sunlight. UV radiation from the sun induces formation of sunburn cells, or apoptotic keratinocytes, in skin.1 UV radiation also induces mutations in the p53 tumor suppressor gene, and inactivation of p53 decreases the frequency of apoptotic cells in UV irradiated mice.1,2 This decreased apoptotic rate in cells with nonfunctional p53 may lead to the survival and expansion of preneoplastic cells, which can accumulate additional genetic damage and progress to cancer. A better understanding of the signaling events which regulate UV-induced apoptosis in keratinocytes is needed to clarify mechanisms of skin photocarcinogenesis.
The signaling pathways which trigger a cell to undergo apoptosis in response to UV radiation are cell type specific and are currently being defined. Activation of the caspase family of cysteine proteases by a variety of apoptotic stimuli, including UV radiation, occurs by proteolytic cleavage and/or clustering of inactive zymogens, and is essential for mediating cell death.3,4,5,6,7 Besides the p53-dependent death pathway noted above, there are two other major signaling pathways described which trigger activation of caspase cascades in response to UV radiation. UV radiation triggers the clustering and activation of many membrane death receptors, including Fas (CD95/APO-1) and tumor necrosis factor receptor, which can in turn activate the initiator caspase-8.3,6,8,9,10 UV radiation also triggers the release of the caspase-9 cofactor cytochrome c from mitochondria. UV-induced cytochrome c release is not inhibited by caspase inhibitors indicating that it is an early event preceding caspase activation.11 Activation of initiator caspases via either mechanism in turn activate effector caspases which cleave a variety of cellular death substrates, including PKCδ.7 Cross-talk exists between the membrane receptor (Fas, TNFR) and mitochondria (cytochrome c) pathways for caspase activation,12,13 however it is unclear how these pathways are interrelated during UV radiation-induced apoptosis.
PKCδ is one of over 40 known caspase substrates, and is cleaved specifically by caspase-3 in vitro.7,14 PKCδ undergoes cleavage in the hinge region (V3) to generate a constitutively active catalytic domain. The cleavage of PKCδ is common to the death effector pathways of diverse apoptotic stimuli, including UV radiation, ionizing radiation, DNA damaging drugs, TNFα, and anti-Fas antibody.5,15,16,17,18 In addition, over-expression of the catalytic domain of PKCδ in several cell types (HeLa, NIH3T3, COS1) is sufficient to induce apoptosis.14,17
We have previously characterized the caspase-dependent activation of PKCδ in normal human keratinocytes undergoing UV-induced apoptosis.5 While other PKC isoforms (PKCε, θ, ζ) can undergo proteolysis by caspases,17,19,20 PKCδ was the only isoform activated by this mechanism in keratinocytes undergoing UV radiation-induced apoptosis.5 We also observed a striking inhibition of UV radiation-induced apoptosis by PKC inhibitors, as assessed by sub-G0 DNA content, DNA fragmentation, and morphological criteria. The induction of apoptosis by over-expression of the PKCδ catalytic domain would not be predicted if this activated form of PKCδ was only responsible for a small subset of apoptotic effects.14,17 Thus we hypothesized that PKC activation may be involved in triggering more upstream events in the apoptotic signaling pathway, such as caspase activation. The current study demonstrates that (i) PKC activity is required for full activation of the caspase cascade, (ii) UV-induced loss of mitochondrial membrane potential requires PKC activity, (iii) the catalytic domain fragment of PKCδ localizes to the mitochondria, and (iv) over-expression of the PKCδ catalytic domain is sufficient to induce apoptosis and loss of mitochondrial membrane potential.
Results
Inhibition of PKC rescues cells from UV-induced cell death
To determine if keratinocytes protected from UV-induced apoptosis by the PKC inhibitor GF 109203X were still metabolically viable, we used the LIVE/DEAD assay to detect metabolically active cells with intact plasma membranes. As shown in Figure 1A, UV irradiation of normal human keratinocytes induced 42% apoptotic cells after 18 h as measured by sub-G0 DNA content, and caused ∼32% cell death as measured by the LIVE/DEAD assay (Figure 1B). Inhibition of PKC with GF 109203X almost completely protected from UV-induced cell death in both the apoptosis and cell viability assay (75 and 84% protection respectively). GF109203X alone had no significant effect on either baseline apoptosis or cell viability, but did significantly protect from UV-induced apoptosis and loss of cell viability (P<0.05).

Protection from UV-induced apoptosis and loss of cell viability by PKC inhibition. Normal keratinocytes (A and B) or HaCaT cells (C and D) were irradiated with UV (30 or 25 mJ/cm2 respectively) in the presence or absence of 5 μM GF 109203X and assayed after ∼18 h. For A and C, cells were stained with propidium iodide, and apoptotic cells with sub-G0 DNA content quantified by flow cytometry. In B and D, cells were assayed for viability with the LIVE/DEAD assay. Note the reversal of UV-induced apoptosis and loss of cell viability by GF 109203X. The data presented is averaged from three independent experiments with error bars denoting standard errors
To determine if PKC activity is required for the induction of apoptosis by UV radiation in cells with mutant p53, we utilized the human keratinocyte cell line HaCaT, which contains inactivating, UV signature mutations in both p53 alleles.21 The p53 tumor suppressor gene is involved in induction of apoptosis by DNA damaging agents such as UV radiation.1 Shown in Figure 1C, HaCaT cells were susceptible to UV-induced apoptosis, and inhibition of PKC activity with GF 109203X significantly reduced their apoptotic response (58% inhibition, P<0.01). HaCaT cell viability was also reduced following UV irradiation (Figure 1D), and inhibition of PKC with GF 109203X blocked this increase in metabolically dead cells to a similar extent (56% inhibition, P<0.05).
To verify that the general PKC inhibitor GF 109203X inhibits the activity of the PKCδ catalytic domain, we performed PKC assays on recombinant PKCδ that had been cleaved by caspase-3. Figure 2A shows the almost complete cleavage of full length PKCδ to a catalytic domain fragment by caspase-3. Figure 2B shows that cleavage with caspase-3 increased the activity of full length PKCδ ∼12.5-fold in the absence of any lipid cofactors. Addition of 100 nM GF 109203X reduced the caspase-3 activated PKCδ activity (96% inhibition).

GF 109203X inhibits the activity of the PKCδ catalytic fragment. (A) Recombinant human PKCδ was incubated with or without recombinant caspase-3 for 30 min. The PKCδ Western blot shows the full length PKCδ (arrow) and the PKCδ catalytic fragment (asterisk) generated by caspase-3 proteolysis. (B) PKC activity of full length or cleaved PKCδ was measured in the presence (+GF) or absence (−GF) of 100 nM GF 109203X. Note the 12.5-fold induction of PKC activity by caspase-3 cleavage and inhibition by GF 109203X. The assay was performed in triplicate and error bars denote standard deviation
Early inhibition of PKC activity is not required to block UV-induced apoptosis
The induction of apoptosis by UV is triggered in part by early signaling events, such as clustering and activation of the cell membrane death receptors Fas/CD95 and tumor necrosis factor receptor.3,6,8,10,22,23 To determine if PKC inhibition was blocking apoptosis by inhibiting any early UV responses, GF 109203X was added to cultured normal human keratinocytes at different times following exposure to UV, and apoptosis assayed 24 h after UV. Shown in Figure 3A, UV radiation-induced apoptosis of normal human keratinocytes was inhibited 62% when GF 109203X was added immediately after UV exposure (0 h), and this inhibition was only slightly diminished when GF 109203X addition was delayed 6 h (55% inhibition). Pretreatment of cells with GF 109203X inhibited UV-induced apoptosis to the same extent as when GF 109203X was added immediately after UV (data not shown), and apoptosis was not inhibited when GF 109203X was added immediately before harvesting the cells for flow cytometry (24 h). These results are consistent with GF 109203X protecting from apoptosis by inhibiting the activity of the PKCδ catalytic domain, which was not detected during the first 8 h after UV exposure.5 In addition, caspase-3-like activity (DEVD substrate) was not induced until 9 h after UV exposure (Figure 3B), again consistent with the kinetics of PKCδ cleavage. Caspase-8-like activity (IETD substrate) was only slightly increased following UV and did not precede caspase-3 activity.

Protection from UV-induced apoptosis does not involve inhibition of death receptor activation. (A) Normal keratinocytes were either untreated or exposed to 30 mJ/cm2 UV, and 5 μM GF 109203X (GF) was added at the time of irradiation (0 h), or 1–24 h after irradiation. Cells were harvested 24 h after UV exposure, and apoptotic cells with sub-G0 DNA content quantified by flow cytometry. Note the equal protection afforded by GF 109203X when added 0–6 h after UV exposure. Data shown is the average of three independent experiments, with error bars denoting standard error. (B) Normal keratinocytes were exposed to 30 mJ/cm2 UV and harvested at the indicated times for caspase activity assay using the caspase-3 substrate (DEVD) or caspase-8 substrate (IETD). Note the progressive increase in caspase-3-like activity after 6 h with significant (P<0.05) increases 9–18 h after UV exposure. Data shown is average of two independent experiments, with error bars denoting the range. (C) Normal keratinocytes were either untreated, treated with 5 μg/ml cyclohexamide for 2 h and then exposed to 100 ng/ml anti-Fas, or exposed to 30 mJ/cm2 UV for 24 h. GF 109203X was added to the cells 15 min before treatments as indicated. Cell were harvested and apoptotic cells quantified by flow cytometry. Note that GF 109203X afforded protection from UV-induced apoptosis, but not from anti-Fas. Data shown is average of three independent experiments, with error bars denoting standard error
We also determined if inhibition of PKC was able to protect from apoptosis induced directly by activation of the membrane death receptor Fas. Figure 3C shows that inhibition of PKC with GF 109203X did not protect from apoptosis induced by anti-Fas plus cyclohexamide, but did block UV-induced apoptosis in the same set of experiments. Taken together, these results indicate that the inhibition of UV-induced apoptosis by PKC inhibition was not due to blocking an early event (i.e. death receptor activation) that precedes the increase in caspase activation and subsequent generation of the PKCδ catalytic domain.
PKC activity is required for full activation of caspases by UV radiation
Since activation of the caspase proteases is a critical component of the UV radiation death effector pathway,3,4,5 we determined if the inhibition of PKC activity could block activation of caspases by UV radiation. We examined the proteolysis of the caspase-3 substrate PKCδ, pro-caspase-3, and pro-caspase-8 in normal keratinocytes. Shown in Figure 4A, UV radiation stimulated the cleavage of PKCδ, pro-caspase-3, and pro-caspase-8 in normal keratinocytes. The PKC inhibitor GF 109203X had no effect on the total levels of the proteins, but GF 109203X did protect from the UV-induced cleavage of PKCδ, pro-caspase-3, and pro-caspase-8.

Inhibition of UV-induced pro-caspase cleavage and activation by PKC inhibition. Normal keratinocytes were irradiated with 30 mJ/cm2 UV and cultured for 18 h in the presence or absence of 5 μM GF 109203X as indicated. For immunoblots (A), proteins were analyzed by SDS–PAGE, and blotted with antibodies to PKCδ, caspase-3, and caspase-8. The arrows indicate full-length proteins, and the asterisks denotes the processed forms of PKCδ, caspase-3, and caspase-8. The lower panels in caspase-3 and caspase-8 are longer exposures which show the cleavage products. Note the UV-induced cleavage of caspase-3, caspase-8, and PKCδ by UV, and inhibition of cleavage by the PKC inhibitor GF 109203X. (B) Lysates were assayed for caspase activity using either the caspase-3 substrate (DEVD) or caspase-8 substrate (IETD). Note the partial reversal of UV-induced caspase activation by the PKC inhibitor GF 109203X. Data shown is the average of three experiments (DEVD) or six experiments (IETD) with error bars denoting standard error
To determine if the inhibition of pro-caspase proteolysis by PKC inhibition corresponds to a decrease in caspase activity, we assayed caspase enzyme activity using substrates for caspase-3 (DEVD-pNA) or caspase-8 (IETD-pNA). In normal keratinocytes, UV induced the activity of both caspases, with caspase-3-like activity increasing ∼fourfold and caspase-8-like activity increasing about ∼1.5-fold (Figure 4B). The UV-induced activities for cleavage of caspase-3 and caspase-8 substrates were inhibited 76 and 62% respectively by the PKC inhibitor GF 109203X. These results indicate that PKC activity is required for the full activation of caspases-3 and -8 following UV radiation.
Expression of PKCδ catalytic domain induces apoptosis
Over-expression of the PKCδ catalytic domain has been demonstrated to induce apoptosis in NIH3T3, HeLa, and COS1 cells.14,17 To determine if expression of the PKCδ catalytic domain was sufficient to induce apoptosis in human keratinocytes, we transiently transfected a human PKCδ catalytic domain construct containing an amino-terminal FLAG epitope tag into normal human keratinocytes and HaCaT cells. Shown in Figure 5, over-expression of the PKCδ catalytic domain in normal human keratinocytes induced nuclear hallmarks of apoptosis. Figure 5A shows propidium iodide stained untreated keratinocytes with round nuclei of uniform staining intensity. After UV radiation exposure, the nuclei of keratinocytes become fragmented and condensed (Figure 5B). Transfection of the PKCδ catalytic domain plasmid triggered condensation and fragmentation of nuclei in cells expressing the catalytic domain of PKCδ (Figure 5C, D). Over-expression of PKCδ catalytic domain in HaCaT cells also induced nuclei fragmentation and chromatin condensation (data not shown).

Expression of PKCδ catalytic domain induces chromatin condensation and nuclear fragmentation in human keratinocytes. Normal keratinocytes were either untreated (A), irradiated with 30 mJ/cm2 UV (B), or transfected with the PKCδ catalytic domain construct, pFLAG-PKCδ-Cat (C and D), and stained with propidium iodide after 18 h (A and B) or 36 h (C and D). Cells were also stained with an anti-FLAG antibody to detect the ectopic PKCδ catalytic domain. Propidium iodide staining is shown in A, B and C, and anti-FLAG staining of cells in C is shown in D. The arrows in C and D indicate PKCδ catalytic domain-expressing cells with chromatin condensation and fragmented nuclei. Note the similar nuclear morphology for UV-irradiated cells (B) and PKCδ catalytic domain-expressing cells (C). For E and F, HaCaT cells were transfected with the vector pFLAG-CMV-2 (E) or pFLAG-PKCδ-Cat (F). After 36 h, cell were fixed and stained with antibodies to the FLAG epitope, followed by FITC-conjugated secondary antibody. Note the characteristic apoptotic cell blebbing and fragmentation in pFLAG-PKCδ-Cat expressing HaCaT cells
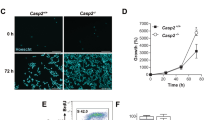
Over-expression of the PKCδ catalytic domain in HaCaT cells caused the formation of many small, round apoptotic bodies which stained strongly with an anti-FLAG antibody (Figure 5F). No FLAG staining was seen in the vector transfected cells (Figure 5E). In addition to many small strongly staining cell fragments, some condensed cells were also seen which morphologically resemble cells undergoing apoptosis. Some FLAG positive cells appeared morphologically normal 36 h after transfection, however almost all cells expressing the PKCδ catalytic domain undergo apoptosis by 72 h (Figure 6). These data, along with the PKC inhibitor data in Figure 1, indicate that PKC-dependent apoptosis does not require wild type p53.

Inhibition of PKCδ catalytic fragment apoptosis by GF 109203X. (A) HaCaT cells were infected with retroviruses encoding either the empty vector (Linker), PKCδ catalytic domain (PKCδ-Cat), or a kinase dead mutant of the PKCδ catalytic domain [PKCδ-Cat (K378A)]. The day after infection, the cells were cultured with or without 5 mM GF 109203X to inhibit PKCδ activity. Shown are phase contrast pictures 3 days after infection. Note the cell loss in the PKCδ-Cat infected cells which is protected by GF 109203X. (B) Immunofluorescence staining of PKCδ one day after infecting HaCaT cells with either the Linker, PKCδ-Cat, or PKCδ-Cat (K378A) virus. Note the elevated staining in the majority of the PKCδ-Cat and PKCδ-Cat (K378A) infected cells. (C) Apoptosis was measured using the TUNEL assay 3 days after HaCaTs were infected with either the Linker, PKCδ-Cat, or PKCδ-Cat (K378A) virus and cultured with (+GF) or without (−GF) 5 μM GF 109203X. Note the induction of apoptosis by the active PKCδ catalytic fragment, but not the inactive K378A mutant, and the inhibition of apoptosis by GF 109203X. The data shown is averaged from three independent experiments and error bars denote standard errors
To determine if the PKC inhibitor GF 109203X could inhibit apoptosis induced by expression of the PKCδ catalytic domain, we infected HaCaT cells with retroviruses encoding either LZRS-Linker vector, PKCδ catalytic domain, or an ATP binding site mutant PKCδ catalytic domain (K378A), and grew the cells with or without 5 μM GF 109203X. Shown in Figure 6A, infection with the catalytically active PKCδ-Cat virus caused massive morphological cell death after 3 days, but the Linker and kinase dead mutant virus did not. Furthermore, GF 109203X protected from this morphological cell death. Infection efficiency was evaluated by immunofluorescence staining cells 1 day after infection, and a low level of over-expression could be detected in 80–90% of the PKCδ-Cat and PKCδ-Cat (K378A) infected cells (Figure 6B). Western analysis indicated that the active and inactive PKCδ catalytic domains were expressed at levels approximating 25% of the full length PKCδ protein (data not shown). As shown in Figure 6C, this modest level of expression was able to induce apoptosis as measured by the TUNEL assay. Apoptosis was induced ∼11-fold by expression of the active PKCδ catalytic domain, but not the inactive mutant, and GF 109203X significantly inhibited this apoptosis (P<0.01). These results indicate that PKCδ catalytic domain enzymatic activity can induce apoptosis, and GF 109203X can block this effect.
Inhibition of PKC protects from UV-induced loss of mitochondria membrane potential
Mitochondrial changes are critical for both the inductive and effector phases of apoptosis.24 Mitochondrial swelling occurs early and is linked to the rupture of the outer mitochondrial membrane and release of cytochrome c. At later time points, the loss of mitochondrial membrane potential signifies metabolic cell death. We have used the mitochondria membrane potential indicator Rhodamine 123 to assess the effects of either PKC or caspase inhibition on the mitochondrial membrane potential of UV-irradiated normal keratinocytes. As shown in Figure 7A, UV irradiation caused a dramatic reduction in Rhodamine 123 staining after 24 h, with 52% of the cells having low mitochondrial membrane potential. The PKC inhibitor GF 109203X was able to almost completely block the UV-induced loss of Rhodamine 123 staining, with only 9.3% of the cells having low mitochondrial membrane potential. The general caspase inhibitor z-VAD also protected keratinocytes from UV-induced loss of mitochondrial membrane potential, but to a lesser extent.

Inhibition of PKC protects keratinocytes from UV-induced loss of mitochondrial membrane potential. (A) Normal human keratinocytes were exposed to 30 mJ/cm2 UV and/or treated with the PKC inhibitor GF 109203X (GF), or the general caspase inhibitor z-VAD as indicated. After 24 h, cells were stained with Rhodamine 123 and mitochondrial membrane potential assayed by flow cytometry. Cells with disrupted mitochondria potential were quantified as having low Rhodamine 123 fluorescence. Note the decrease in Rhodamine 123 staining in the UV exposed cells which was inhibited by GF 109203X, and to a lesser extent by z-VAD. Similar results were obtained in three additional experiments. (B) Normal keratinocytes were exposed to 30 mJ/cm2 UV in the presence or absence of 5 μM GF 109203X and assayed for viability after ∼18 h using the MTT assay. Note the protection from UV-induced cell death by the PKC inhibitor GF 109203X. Data is the average from three experiments, with error bars denoting standard error. (C) HaCaT cells were infected with viruses encoding either vector (Linker), PKCδ catalytic domain (PKCδ-Cat), or the inactive mutant [PKCδ-Cat (K378A)], and then cultured with (+GF) or without (−GF) 5 mM GF 109203X. After 3 days, mitochondrial membrane potential was assayed by Rhodamine 123 staining and flow cytometry. Note the loss of Rhodamine 123 staining by the active PKCδ catalytic domain virus, and protection by GF 109203X. Data is the average from three experiments, with error bars denoting standard error
We also used the MTT cell viability assay as a measure of mitochondrial function since viability in this assay is dependent upon mitochondrial redox reactions. UV radiation caused a 41% decrease in cell viability as measured by the MTT assay and GF 109203X was able to almost completely restore cell viability (Figure 7B). The protection from UV-induced mitochondrial damage by GF 109203X indicates that UV-induced loss of mitochondrial function is dependent upon PKC activation.
To determine if the active PKCδ catalytic domain was sufficient to disrupt mitochondrial membrane potential, we infected HaCaT cells with the vector control Linker, active PKCδ-Cat, or inactive PKCδ-Cat (K378A) viruses and assayed mitochondrial membrane potential 3 days later. Figure 7C shows that the PKCδ catalytic domain virus caused significant (P<0.05) loss of mitochondrial membrane potential as measured by loss of Rhodamine 123 staining. GF 109203X was able to completely block this loss of mitochondrial staining, and the kinase dead mutant virus was not significantly different than the control Linker virus. These results demonstrate that expression of the catalytically active PKCδ catalytic domain is sufficient to disrupt mitochondrial membrane potential.
The PKCδ catalytic domain is localized to mitochondria
To determine if PKC inhibition blocks the UV radiation-induced release of cytochrome c from the mitochondria, we prepared mitochondria-enriched and cytosol fractions from keratinocytes for immunoblot analysis. Figure 8A shows that cytochrome c was predominately in the mitochondrial fraction in untreated cells (94% mitochondrial), but was released into the cytosol 24 h after UV irradiation (54% mitochondrial). The PKC inhibitor GF 109203X did not prevent the release of cytochrome c from the mitochondria-enriched fraction (47% mitochondrial). These same fractions were also immunoblotted for PKCδ, caspase-8 and caspase-3 to determine the relative distribution of these proteins. In untreated cells, PKCδ was largely in the cytosol fraction, but translocated to the mitochondrial fraction following UV irradiation. In fact, the catalytic domain of PKCδ was detected exclusively in this mitochondrial fraction. PKC inhibition did not block the translocation of PKCδ, but did prevent the cleavage, as previously observed in whole cell lysates (Figure 4A). PKC inhibition also blocked the appearance of the caspase-8 cleavage product, which was localized to the mitochondrial fraction. Pro-caspase-3 and its cleavage product were predominately detected in the cytoplasmic fraction, and the generation of the caspase-3 cleavage product was again inhibited by PKC inhibition. To determine the subcellular localization of transfected PKCδ catalytic domain, we transfected pFLAG-PKCδ-Cat into HaCaT cells and fractionated them into mitochondrial and cytosol fractions. Ectopically expressed PKCδ catalytic domain was also localized predominately in the mitochondrial fraction in transfected HaCaT cells, as was the endogenous PKCδ catalytic domain induced by UV radiation (Figure 8B). The reason for the slightly slower migration of the ectopic PKCδ catalytic fragment compared to the endogenous fragment is unknown, but may be due to the FLAG epitope or additional cleavage events in UV irradiated cells.

Effect of PKC inhibition on mitochondrial localization of cytochrome c and PKCδ. (A) Normal keratinocytes were left untreated, or exposed to 30 mJ/cm2 UV with or without 5 μM GF 109203X (GF) and cells fractionated into mitochondria-enriched (M) and cytosol (C) fractions after 24 h. Shown in (A) are immunoblots for cytochrome c, PKCδ, caspase-8 and caspase-3. The bottom panel is a longer exposure of caspase-3 that shows cleavage products. Note the release of cytochrome c from the mitochondria by UV, and the recruitment of PKCδ catalytic domain to the mitochondrial fraction. Similar results were obtained in a total of four experiments. (B) HaCaT cells were transfected with the pFLAG-CMV-2 vector (pCMV-2), pFLAG-PKCδ-Cat (PKCδ-Cat), or exposed to 30 mJ/cm2 UV and fractionated into mitochondria-enriched (M) and cytosol (C) fractions. Shown in B is an immunoblot for PKCδ. Note the localization of ectopically expressed PKCδ catalytic domain at ∼42 kDa in the mitochondria-enriched fraction
To further characterize the subcellular localization of the PKCδ catalytic domain, we performed confocal microscopy on HaCaT cells transfected with the PKCδ catalytic domain. As shown in Figure 9, HaCaT cell mitochondria stained with the mitochondrial probe MitoTraker Red are distributed in a punctate, perinuclear pattern in most cells. Two-color fluorescence reveals that the transfected PKCδ catalytic domain largely co-localizes with the mitochondria, as indicated by the yellow fluorescence in Figure 9C.

Localization of PKCδ catalytic domain to mitochondria by confocal microscopy. HaCaT cells were transfected with pFLAG-PKCδ-Cat and stained with Mito Traker Red and anti FLAG antibody after 24 h. Mito Traker staining is red and labels the mitochondria (A), FLAG epitope staining is green and shows the PKCδ catalytic domain (B), and the merged image in (C) shows co-localization in yellow. Bars denote 20 microns
Discussion
The induction of apoptosis by UV radiation in keratinocytes can be largely reduced by inhibition of PKC activity (Figures 1 and 3).5,25 This inhibition also occurred in HaCaT cells which have mutant p53, indicating that the PKC-dependent apoptosis does not require wild type p53. The p53 independence of PKCδ-induced apoptosis was further supported by the induction of apoptosis in HaCaT cells over-expressing the PKCδ catalytic domain (Figures 5 and 6). Despite their mutant p53 status, HaCaT cells do undergo apoptosis in response to UV irradiation (Figure 1),3,8 indicating that other p53-independent apoptotic pathways exist in these cells.
The PKC inhibitor GF 109203X was able to block UV-induced apoptosis even when it was added 6 h after UV exposure, indicating that PKC activation is not critical in early apoptotic signaling (Figure 3A). UV activates multiple signaling pathways which contribute to apoptosis, including the rapid activation (5–45 min) of membrane death receptors (Fas, TNFR).2,3,6,8,10,26 In addition, Chen et al. detected translocation (5–30 min) of PKCδ and PKCε to the membrane following UV exposure suggesting rapid activation.25 Our results (Figure 3) indicate that inhibition of PKC activation at these early times is not required to block apoptosis. Furthermore, PKC inhibition was unable to protect from apoptosis induced directly by anti-Fas (Figure 3C), suggesting that PKC activation may be more essential for the intrinsic death pathway initiated by UV-induced DNA damage27 than membrane receptor-initiated death signaling.
Inhibition of PKC activity also protected from the loss of cell viability induced by UV radiation (Figures 1 and 7B). Cell viability was measured by the LIVE/DEAD assay, which is dependent on an intact plasma membrane and cellular esterase activity. Inhibition of PKC activity also protected from the loss of cell viability as measured by the MTT assay, which is dependent upon mitochondrial redox reactions (Figure 7B). Consistent with PKC activation being involved in disrupting mitochondrial function, we found that inhibition of PKC activity also protected cells from the loss of mitochondrial membrane potential (Figure 7). Loss of mitochondrial membrane potential is a late event in UV radiation-induced apoptosis which can be blocked by caspase inhibitors (Figure 7A).11 Since PKC inhibition also blocked caspase activation (Figure 4), the protection from mitochondria dysfunction as measured by the MTT assay and Rhodamine 123 staining may be due to inhibition of caspase activation by GF 109203X.
Inhibition of PKC activity reduced the UV-stimulated proteolytic processing of PKCδ, pro-caspase-3, and pro-caspase-8 in normal keratinocytes (Figures 4 and 8). The activities of caspase-3 and caspase-8 were also suppressed by PKC inhibition, as measured by in vitro enzyme assays using peptide substrates, and in cells as measured by cleavage of endogenous substrates PKCδ and pro-caspase-3 (Figure 4).14,28 The inhibition of pro-caspase-8 processing by PKC inhibition suggests PKC is an essential component for triggering the caspase cascade, in addition to its previously well characterized role as a caspase-3 substrate.14,16,17 These results are in agreement with other investigators who have found that inhibition of PKCδ with rottlerin inhibited caspase-3 activation and PKCδ cleavage during chemotherapy drug-induced apoptosis in HeLa and salivary gland acinar cells.18,29 In HeLa cells, doses of Bryostatin 1 which down-regulated PKCδ, also blocked caspase-3 and caspase-7 activation, however the activation of upstream caspases was not examined.29 Rottlerin is reported to be a PKCδ - selective inhibitor in vitro,30 and also blocked UV radiation-induced apoptosis in JB6 mouse epidermal cells.25 We have seen inhibition of UV-induced apoptosis in keratinocytes by rottlerin, however rottlerin alone caused cell toxicity as determined by LIVE/DEAD and MTT cell viability assays (data not shown).
Our current model for the kinetics of PKCδ cleavage and activation in UV apoptosis places PKCδ as a critical enhancer of caspase activation. Within minutes, UV activates membrane death receptors and causes DNA damage, but it takes over 6 hours before caspase-3 becomes active and begins to cleave PKCδ to generate the active catalytic fragment (Figure 3B).5 The active PKCδ translocates to the mitochondria where it disrupts mitochondrial membrane potential and greatly potentiates caspase activation, setting up a positive feedback loop between caspase activation and PKCδ cleavage/activation. This model explains why inhibition of PKC activity can be delayed 6 h after UV exposure, and why inhibition of PKC or caspase activity blocks cleavage of both PKC and caspases. An alternative model is that PKC is activated before the caspases by a proteolysis-independent mechanism, which then triggers the initiation of the caspase cascade and subsequent PKCδ cleavage. This model is unlikely because the inhibition of PKC prior to caspase activation does not afford significantly greater protection from UV-induced apoptosis than with the continued presence of the PKC inhibitor (Figure 3A). In addition, we are unable to detect PKC activity at time points preceding caspase activation/PKCδ cleavage (data not shown), but we can detect PKC activation after caspase activation/PKCδ cleavage.5 Finally, the addition of the PKC inhibitor GF 109203X 6 h after UV is still able to block caspase-3 activation (data not shown). Taken together, our data strongly supports positive feedback regulation between PKCδ and caspases during UV-induced apoptosis.
Over-expression of full length PKCδ in mouse and human keratinocytes or human LNCaP prostate cancer cells greatly enhanced the induction of apoptosis triggered by the phorbol ester TPA.31,32 In keratinocytes, the mechanism of apoptosis induced by full length PKCδ appears to be similar to PKCδ-catalytic domain-mediated apoptosis since both proteins are localized to the mitochondria, their activation is associated with loss of mitochondrial function, and they are involved in caspase-3 activation (Figures 4, 7, 8 and 9).31 However in LNCaP cells, PKCδ-induced apoptosis does not involve caspase-3 activation, indicating cell-type specific caspase regulation.32 Differences in caspase involvement for the induction of apoptosis in different cell types has been previously observed between caspase-9 deficient fibroblasts and thymocytes.33 In addition, pre-treatment of human keratinocytes with the PKC activator TPA triggered growth arrest and reduced the UV-induced apoptosis, indicating that the timing of PKC activation can influence the sensitivity of cells to apoptotic stimuli.34
Over-expression of the catalytic domain of PKCδ was sufficient to induce apoptosis in normal keratinocytes and HaCaT cells (Figures 5 and 6). Ultrastructural hallmarks of apoptosis could also be identified in HaCaT cells transfected with the PKCδ catalytic domain by transmission electron microscopy (data not shown). Since the catalytic domain of PKCδ is induced by UV radiation in keratinocytes, the constitutively active PKCδ catalytic domain may contribute to the UV radiation apoptotic phenotype. While the catalytic domains of other PKC isoforms (PKCε, PKCθ, PKCζ) are generated by apoptotic stimuli in other cell types, and may also be able to stimulate apoptosis,17,19,29 PKCδ is the only PKC isoform cleaved in keratinocytes exposed to UV radiation.5
We also found that UV triggered the translocation of the PKCδ catalytic domain to the mitochondria (Figure 8). A mitochondrial role for active forms of PKCδ in stimulating apoptosis is supported by several observations; (i) the activated full length PKCδ and catalytic fragment of PKCδ generated after UV exposure are localized to the mitochondria (Figures 8 and 9),31,35 (ii) activation of PKCδ is associated with loss of mitochondrial membrane potential (Figure 7);31 (iii) expression of the active, but not inactive, PKCδ catalytic fragment can trigger loss of mitochondrial membrane potential (Figure 7C); and (iv) PKCδ-induced apoptosis can be blocked by Bcl-2 over-expression, which is a mitochondrial-localized protein.24,32 While the biochemical role of the PKC δ catalytic domain in UV-induced apoptosis is currently unclear, release of cytochrome c from the mitochondria in response to UV does not require PKC activity since its release was not blocked by GF 109203X (Figure 8). A recent report demonstrates that TPA stimulates the release of cytochrome c from mitochondria in U-937 cells, and that this can be blocked by rottlerin or a kinase dead PKCδ.35 The activities of several Bcl-2 family members are regulated by phosphorylation,36,37 and these mitochondrial proteins are attractive candidates for PKCδ catalytic domain substrates.
In summary, we have identified a positive feedback loop involved in amplifying the apoptotic response of human keratinocytes to UV radiation. Protein kinase C plays a dual role in this pathway, being required for full activation of caspases, and also being activated directly by caspase proteolysis.5 Since PKCδ is a widely expressed PKC isoform which is cleaved in a variety of cell type undergoing apoptosis, this dual role for PKC is likely to be a general pathway functioning to accelerate apoptosis of cells committed to die.5,16,17,29,38 In addition, we have localized the catalytic domain of PKCδ generated in response to apoptotic stimuli to the mitochondria, and have identified a functional role of PKC activity in disruption of mitochondrial function in UV irradiated keratinocytes. These results have relevance for skin carcinogenesis, as loss or inactivation of PKCδ in keratinocytes with activated ras39,40 may allow these premalignant cells to resist apoptosis and promote the emergence of malignant clones.
Materials and Methods
Antibodies and chemicals
PKCδ was detected using an antibody raised against a carboxy-terminal peptide (sc-937, Santa Cruz Biotech., Inc. Santa Cruz, CA, USA) at 1 : 4000 for immunoblotting, and at 1 : 100 for immunofluorescence. The FLAG epitope was detected with the M2 monoclonal antibody at 1 : 100 for immunofluorescence (F-3165, Sigma Chemical Co. St. Louis, MO, USA). Other antibodies used for immunoblotting were anti-caspase-3 at 1 : 200 (sc-7272, Santa Cruz Biotech., Inc.), anti-human caspase-8 (FLICE) at 1 : 1000 (05-477, Upstate Biotech., Lake Placid, NY, USA), and anti-cytochrome c at 1 : 500 (sc-7159, Santa Cruz Biotech.). The activating anti-Fas antibody (clone CH11) was used at 100 ng/ml (05-201, Upstate Biotech.).
The PKC inhibitor GF 109203X (Bisindolylmaleimide I), which is competitive for ATP and can inhibit the activity of free PKCδ catalytic domain, was purchased from Alexis Biochemicals (San Diego, CA, USA). GF 109203X was routinely used at 5 μM, but had an ED50 for inhibition of UV-induced apoptosis of 0.85 μM (data not shown). MitoTraker Red CMXRos and Rhodamine 123 were purchased from Molecular Probes (Eugene, OR, USA) and used at 100 nM and 5 μM respectively. For anti-Fas experiments, cells were pretreated with 5 μg/ml cyclohexamide (Sigma Chemical Co., St. Louis, MO, USA) for 2 h before addition of anti-Fas antibody. Cyclohexamide pretreatment was necessary to get significant induction of apoptosis in keratinocytes. The colorimetric caspase substrates Ac-DEVD-pNA and Ac-IETD-pNA, and the caspase inhibitor z-VAD-FMK were purchased from Enzyme Systems Products (Livermore, CA, USA).
Caspase and PKC assays
Caspase activity was assayed as described by Takahashi et al. with minor modifications.41 Following treatments, the cells were washed twice with ice cold PBS and lysed in caspase lysis buffer (25 mM HEPES, 10% sucrose, 0.1% CHAPS, 2 mM EDTA). The lysates were spun in a microcentrifuge for 3 min at 4°C, and the supernatant removed for assaying. Equal amounts of protein (150–300 μg) were diluted to 50 μl with caspase lysis buffer, and incubated with an equal volume of 25 mM HEPES, 5 mM DTT and 100 μM colorimetric caspase substrate (Ac-DEVD-pNA or AC-IETD-pNA) at 37°C for 1–4 h. Absorbance at 405 nm was measured, and nmoles of cleaved pNA substrate was calculated from a standard curve.
PKC assays were performed with purified full length PKCδ or caspase-3-cleaved PKCδ. Prior to measuring PKC activity, 200 ng of recombinant human PKCδ (PanVera Corp., Madison, WI, USA) was incubated with 400 ng of purified caspase-3 (Alexis Biochemicals) for 30 min at 37°C in 160 μl of 25 mM HEPES, 0.25 M sucrose, 1 mM EDTA, 2.5 M DTT to cleave and activate PKCδ. Cleavage was verified by Western blotting. PKC activity was then measured in 50 μl reactions containing 50 mM Tris-HCl, pH 7.4, 250 μg/ml BSA, 1 mM EGTA, 10 μM Ser25 peptide substrate (Life Technologies Inc. Gaithersburg, MD, USA), 1.5 mM MgCl2, and 25 ng of full length or cleaved PKCδ. Reactions were started by adding 25 μM ATP containing 1 μCi [γ-32P]ATP, and incubated for 10 min at 30°C. Reactions were stopped by spotting 25 μl on P81 phosphocellulose disks, washing extensively in 0.45% phosphoric acid, once in acetone, and counting the associated radioactivity in a scintillation counter. One hundred nM GF 109203X was added to some assays to inhibit PKC activity.
Cell culture and UV treatment
Normal human epidermal keratinocytes were isolated from neonatal foreskins following routine circumcision as previously described.5,42,43 The immortalized human keratinocyte cell line HaCaT, which has two mutant p53 alleles,21 was kindly provided by Dr Norbert Fusenig (German Cancer Research Center, Heidelberg, Germany).44 All cells were cultured either in KGM (Clonetics Corp., San Diego, CA, USA) or Media 154CF with 0.15 mM calcium added (Cascade Biologics, Inc., Portland, OR, USA).
Keratinocytes were irradiated with dish lids removed by a Panelite Unit (Ultralite Enterprises, Inc, Lawrenceville, GA, USA) containing four UVB bulbs (FS36T12/UVB-VHO). The output wavelengths of the bulbs are 65% UVB, 34% UVA and 1% UVC. The UV dose was monitored with an International Light Inc. (Newburyport, MA, USA) radiometer fitted with a UVB detector. Twenty-five to 30 mJ/cm2 UVB was routinely used for induction of apoptosis.
Cell fractionation and immunoblotting
Total cell lysates for immunoblotting were by prepared by lysing floating and attached cells in 20 mM Tris-HCl, pH 7.5, 1% Triton X-100, 5 mM EDTA, 40 μg/ml leupeptin, 1 μM pepstatin, 1 mM AEBSF. The lysates were spun in a microcentrifuge for 10 min at 4°C, and 30 μg protein from the supernatant taken for SDS–PAGE. For mitochondria enrichment, the cells were suspended in 0.5 ml of ice cold 20 mM HEPES, pH 7.4, 250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2 1 mM EGTA, 1 mM DTT, and Complete Protease Inhibitor Cocktail (Boehringer Mannheim, Mannheim, Germany), and gently homogenized with 30 strokes of a glass homogenizer using a B pestle.45 Unlysed cells and nuclei were pelleted by spinning for 10 min at 750×g. The supernatant was removed and spun at 10 000×g for 25 min at 4°C. The pellet was suspended in lysis buffer and taken as the mitochondria-containing fraction, and the supernatant was spun at 100 000×g for 1 h at 4°C. The supernatant from this spin was taken as the cytosol fraction. Twenty μg of protein from the cytosol and mitochondrial fractions were used for immunoblotting.
The proteins were run on SDS polyacrylamide gels, transferred to nitrocellulose, and stained with Ponceau S to ensure equal protein loading. The membranes were blocked with 5% milk in 20 mM Tris-HCl, pH 7.6, 150 mM NaCl (TBS), stained with primary antibody and horseradish peroxidase-conjugated secondary antibody for 1 h each, and washed extensively in TBS, 0.05% Tween 20. Proteins were detected using the ECL detection kit (Amersham Life Sciences, Piscataway, NJ, USA).
DNA constructs
For construction of the PKCδ catalytic domain expression vector pFLAG-PKCδ-Cat, the full length human PKCδ cDNA (80048, American Type Culture Collection, Manassas, VA, USA) was first excised with NheI and cloned into the XbaI site of pBluescript KS (Stratagene, La Jolla, CA, USA). A clone with the 5′ end of PKCδ oriented closest to the SacI site of the multiple cloning site was designated pKS-PKCδ-2. The catalytic domain of PKCδ was excised from pKS-PKCδ-2 with BglII/BamHI digestion, and cloned into the complimentary BglII/BamHI sites of pFLAG-CMV-2 (Sigma Chemical Co.). This generated pFLAG-PKCδ-Cat, which contains the entire catalytic domain of PKCδ starting 7 amino acids carboxy-terminal to the caspase-3 cleavage site, and an amino-terminal FLAG epitope tag. Transfection of pFLAG-PKCδ-Cat into COS7 cells caused an increase in basal PKC activity, indicating that our construct produced enzymatically active protein (data not shown).
For construction of the retroviral expression vectors, a C-terminal FLAG epitope was first introduced into the full length human PKCδ cDNA by PCR. The template plasmid was pKS-PKCδ-1, which contains the PKCδ cDNA in the opposite orientations as pKS-PKCδ-2. The 5′ PCR primer used was MFD9: CACCAAGGAGTCCAAGGACATC, and the 3′ primer was MFD10: ATTGTTTAGCGGCCGCCTACTTGTCCTCGTCGTCCTTGTAATCTTCCAGGAGGTGCTCGAA. The 355 bp product was digested with XcmI/NotI and ligated into XcmI/NotI digested pKS-PKCδ-1. This new construct was named pKS-PKCδ-FLAG. The regulatory domain of PKCδ was removed by EcoRI/BglII digestion, and the gap repaired with a double stranded oligonucleotide encoding a consensus Kozak sequence, initiation methionine, and the 7 amino acids between the caspase-3 cleavage site and the BglII site to generate the entire PKCδ catalytic fragment (pKS-PKCδ-Cat-FLAG). The oligonucleotides used to make this repair were MFD17: AATTCACCATGAACAGTGGGACCTACGGCAA, and MFD18: GATCTTGCCGTAGGTCCCACTGTTCATGGTG, with the initiation methionine underlined. The catalytic domain of PKCδ was excised from pKS-PKCδ-Cat-FLAG with EcoRI/NotI digestion, and subcloned into the EcoRI/NotI sites of the episomal retroviral vector LZRS-Linker.46 LZRS-Linker is a modified version of LZRS-EGFP, provided by Dr Paul Khavari (Stanford University School of Medicine, Stanford, CA, USA), in which EGFP was removed by BamHI/NotI digestion and unique cloning sites (BamHI, HindIII, EcoRI, NotI) introduced by double stranded oligonucleotides. For generation of the kinase inactive catalytic domain, the conserved lysine in the ATP binding site of PKCδ (K378) was mutated to alanine. Site-directed mutagenesis was performed on pKS-PKCδ-2 using the Transformer Site Directed Mutagenesis Kit (Clontech). The selection primer was MFD3: GGCTGCAGGAcgTCGATATCAAGCTTAT and the mutagenesis primer MFD4: AGAGTACTTGCCATCgcGGCCCTCAA, with lowercase letters denoting mutated nucleotides. The BsmI/BglII fragment containing the mutation was swapped with the wild type sequence in pKS-PKCδ-FLAG to generate pKS-PKCδ-FLAG(K378A). This mutant plasmid was then used to generate LZRS-PKCδ-Cat (K378A) in the same manner as the wild type pKS-PKCδ-FLAG. All PCR products and oligonucleotides insertions, including site-directed mutations, were verified by sequencing. The final vectors, named LZRS-PKCδ-Cat-FLAG and LZRS-PKCδ-Cat-FLAG (K378A) were transfected into the Phoenix Ampho retroviral packaging line (ATCC, with permission from Dr Garry P Nolan, Stanford University Medical Center) and used to make retroviruses as described previously.47 Briefly, after calcium phosphate transfection, packaging cells were selected and expanded in the presence of 1 μg/ml puromycin, and virus was harvested from confluent dishes cultured for 24–48 h at 32°C.
Transfections and infections
DNA was transfected into keratinocytes using the FuGene-6 Transfection Reagent (Boehringer Mannheim) according to the manufacturer's protocol. Cells were analyzed 24–48 h after transfection. For infections, cells were plated in six well dishes at 105 per well, and the following day viral supernatant added in the presence of 4 μg/ml polybrene (Hexadimethrine Bromide, Sigma Chemical Co.). The cells were infected by spinning the plates at 300×g for 1 h at 32°C, and the supernatants replaced with fresh Media 154. Cells were infected twice, once in the morning and once in the evening, because the virus was of low titer due to killing of the packaging cells by the active PKCδ catalytic domain. The day after infection, cells were washed three times with PBS- and fed with Media 154 with or without GF 19203X. For protein analysis, cells were harvested the day after plating. For TUNEL and Rhodamine 123 staining, cells were assayed 3 days after infection.
Immunostaining
For immunofluorescence, cells were grown on glass coverslips, and after transfection or treatment, washed with PBS and fixed in −20°C acetone/methanol (1 : 1) for 10 min. Cells were stained with primary antibody diluted in PBS with normal goat serum (1 : 20 dilution) for 1 h at room temp, washed with FA Buffer (Difco Labs., Detroit, MI, USA), and incubated with secondary FITC conjugated antibody at 1 : 40 dilution. Cells were washed in FA Buffer and mounted in 40% polyvinyl alcohol (300–70 000 MW) in glycerol containing 100 mg/ml 1,4 diazabicyclo octane to reduce fading of the fluorescence. For propidium iodide counterstaining, 3 μg/ml propidium iodide was added to the mounting media. For routine immunofluorescence, the cells were viewed with an Olympus AX80 fluorescence microscope. For confocal microscopy, cells were imaged with a Zeiss LSM-510 laser scanning microscope set at 0.8 μm optical slice.
TUNEL assays
Apoptosis was measured in some experiments by the TUNEL assay using the ApopTag Peroxidase Kit (Oncor, Gaithersburg, MD, USA) following the manufacturer's instructions. Briefly, 3 days after infection the cells were trypsinized, combined with floating cells and spun onto glass slides. After air drying for 10–15 min, the cytospins were stored at 4°C until stained. After TUNEL staining, the cells were counterstained with hematoxylin, and the percentage of TUNEL positive cells counted in a blinded fashion.
Flow cytometry
Apoptosis was routinely measured by determining DNA content of cells by propidium iodide staining and flow cytometry, as previously described.5 Briefly, cells were trypsinized, fixed with ethanol, and stained with propidium iodide before being analyzed on a Coulter Epics XL-MCL flow cytometer. Cells with DNA content less than the G0/G1 amount of untreated cells were considered apoptotic.
Mitochondria membrane potential was measured by Rhodamine 123 fluorescence. Cells were trypsinized and incubated for 20 min in 1 ml of room temperature media containing 5 μM Rhodamine 123. The cells were then washed and analyzed on a Coulter Epics XL-MCL flow cytometer for reduced Rhodamine 123 fluorescence, indicating loss of mitochondria membrane potential.
Viability assays
Cell viability was assayed with the LIVE/DEAD Viability/Cytotoxicity Kit (Molecular Probes, Eugene, OR, USA), which uses two mutually exclusive fluorescent dyes to measure cell viability and death based upon intracellular esterase activity and plasma membrane integrity. After treatments, cells were incubated with 0.1 μM calcein AM and 4 μM ethidium homodimer for 30 min at room temperature. Cells were trypsinized, washed with PBS, and resuspended in PBS for flow cytometry. Dead cells were determined as the percentage of singly positive cells which were positive for ethidium. Cell viability was also assayed with the MTT Assay (Boehringer Mannheim, Indianapolis, IN, USA), which is a colorimetric assay based upon reduction of the tetrazolium salt MTT to purple formazan, as recommended by the manufacturer.
Abbreviations
- UV:
-
ultraviolet
- PKC:
-
protein kinase C
References
Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, Brash DE . 1994 Sunburn and p53 in the onset of skin cancer Nature 372: 773–776
Kraemer KH . 1997 Sunlight and skin cancer: another link revealed Proceedings of the Natl. Acad. Sci. USA 94: 11–14
Aragane Y, Kulms D, Metze D, Wilkes G, Poppelmann B, Luger TA, Schwarz T . 1998 Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO-1) independently of its ligand CD95L J. Cell Biol. 140: 171–182
Takahashi H, Kinouchi M, Iizuka H . 1997 Interleukin-1 beta-converting enzyme and CPP32 are involved in ultraviolet B-induced apoptosis of SV40-transformed human keratinocytes Biochem. Biophys. Res. Commun. 236: 194–198
Denning MF, Wang Y, Nickoloff BJ, Wrone-Smith T . 1998 Protein kinase Cδ is activated by caspase-dependent proteolysis during ultraviolet radiation-induced apoptosis of human keratinocytes J. Biol. Chem. 273: 29995–30002
Sheikh MS, Antinore MJ, Huang Y, Fornace AJ Jr . 1998 Ultraviolet-irradiation-induced apoptosis is mediated via ligand independent activation of tumor necrosis factor receptor 1 Oncogene 17: 2555–2563
Thornberry NA, Lazebnik Y . 1998 Caspases: Enemies within Science 281: 1312–1316
Schwarz A, Bhardwaj R, Aragane Y, Mahnke K, Riemann H, Metze D, Luger TA, Schwarz T . 1995 Ultraviolet-B-induced apoptosis of keratinocytes: evidence for partial involvement of tumor necrosis factor-alpha in the formation of sunburn cells J. Invest. Dermatol. 104: 922–927
Rosette C, Karin M . 1996 Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors Science 274: 1194–1197
Rehemtulla A, Hamilton CA, Chinnaiyan AM, Dixit VM . 1997 Ultraviolet radiation-induced apoptosis is mediated by activation of CD-95 (Fas/APO-1) J. Biol. Chem. 272: 25783–25786
Bossy-Wetzel E, Newmeyer DD, Green DR . 1998 Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization EMBO J. 17: 37–49
Kuwana T, Smith JJ, Muzio M, Dixit V, Newmeyer DD, Kornbluth S . 1998 Apoptosis induction by caspase-8 is amplified through the mitochondrial release of cytochrome c J. Biol. Chem. 273: 16589–16594
Li H, Zhu H, Xu CJ, Yuan J . 1998 Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis Cell 94: 491–501
Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, Pandey P, Datta R, Huang Y, Kharbanda S, Allen H, Kamen R, Wong W, Kufe D . 1996 Proteolytic activation of protein kinase C delta by an ICE/CED 3- like protease induces characteristics of apoptosis J. Exp. Med. 184: 2399–2404
Emoto Y, Kisaki H, Manome Y, Kharbanda S, Kufe D . 1996 Activation of protein kinase C delta in human myeloid leukemia cells treated with 1-beta-D-arabinofuranosylcytosine Blood 87: 1990–1996
Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong WW, Kamen R, Weichselbaum R, Kufe D . 1995 Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells EMBO J. 14: 6148–6156
Mizuno K, Noda K, Araki T, Imaoka T, Kobayashi Y, Akita Y, Shimonaka M, Kishi S, Ohno S . 1997 The proteolytic cleavage of protein kinase C isotypes, which generates kinase and regulatory fragments, correlates with Fas- mediated and 12-O-tetradecanoyl-phorbol-13-acetate-induced apoptosis Eur. J. Biochem. 250: 7–18
Reyland ME, Anderson SM, Matassa AA, Barzen KA, Quissell DO . 1999 Protein kinase C delta is essential for etoposide-induced apoptosis in salivary gland acinar cells J. Biol. Chem. 274: 19115–19123
Datta R, Kojima H, Yoshida K, Kufe D . 1997 Caspase-3-mediated cleavage of protein kinase C theta in induction of apoptosis J. Biol. Chem. 272: 20317–20320
Frutos S, Moscat J, Diaz-Meco MT . 1999 Cleavage of zetaPKC but not lambda/iotaPKC by caspase-3 during UV-induced apoptosis J. Biol. Chem. 274: 10765–10770
Lehman TA, Modali R, Boukamp P, Stanek J, Bennett WP, Welsh JA, Metcalf RA, Stampfer MR, Fusenig N, Rogan EM, Harris CC . 1993 p53 mutations in human immortalized epithelial cell lines Carcinogenesis 14: 833–839
Hill LL, Ouhtit A, Loughlin SM, Kripke ML, Ananthaswamy HN, Owen-Schaub LB . 2000 Fas ligand: A sensor for DNA damage critical in skin cancer etiology Science 285: 898–900
Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH . 1998 p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs J. Exp. Med. 188: 2033–2045
Green DR, Reed JC . 1998 Mitochondria and apoptosis Science 281: 1309–1312
Chen N, Ma W, Huang C, Dong Z . 1999 Translocation of protein kinase Cepsilon and protein kinase Cdelta to membrane is required for ultraviolet B-induced activation of mitogen-activated protein kinases and apoptosis J. Biol. Chem. 274: 15389–15394
Leverkus M, Yaar M, Gilchrest BA . 1997 Fas/Fas ligand interaction contributes to UV-induced apoptosis in human keratinocytes Exp. Cell Res. 232: 255–262
Kulms D, Poppelmann B, Yarosh D, Luger TA, Krutmann J, Schwarz T . 1999 Nuclear and cell membrane effects contribute independently to the induction of apoptosis in human cells exposed to UVB radiation Proc. Natl. Acad. Sci. USA 96: 7974–7979
Cohen GM . 1997 Caspases: the executioners of apoptosis Biochem. J. 326: 1–16
Basu A, Akkaraju G . 1999 Regulation of caspase activation and cis-Diamminedichloroplatinum(II)-induced cell death by protein kinase C Biochemistry 38: 4245–4251
Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F . 1994 Rottlerin, a novel protein kinase inhibitor Biochem. Biophys. Res. Commun. 199: 93–98
Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH . 1999 Protein kinase C delta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector Mol. Cell Biol. 19: 8547–8558
Fujii T, Garcia-Bermejo ML, Bernabo JL, Caamano J, Ohba M, Kuroki T, Li Yuspa SH, Kazanietz MG . 2000 Involvement of protein kinase C delta (PKCdelta) in phorbol ester-induced apoptosis in LNCaP prostate cancer cells. Lack of proteolytic cleavage of PKCdelta J. Biol. Chem. 275: 7574–7582
Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia AXd1PJ, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW . 1998 Differential requirement for caspase 9 in apoptotic pathways in vivo Cell 94: 339–352
Chaturvedi V, Qin JZ, Denning MF, Choubey D, Diaz MO, Nickoloff BJ . 1999 Apoptosis in proliferating, senescent, and immortalized keratinocytes J. Biol. Chem. 274: 23358–23367
Majumder PK, Pandey P, Sun X, Cheng K, Datta R, Saxena S, Kharbanda S, Kufe D . 2000 Mitochondrial translocation of protein kinase C delta in phorbol ester-induced cytochrome c release and apoptosis J. Biol. Chem. 275: 21793–21796
Haldar S, Basu A, Croce CM . 1998 Serine-70 is one of the critical sites for drug-induced Bcl2 phosphorylation in cancer cells Cancer Res. 58: 1609–1615
Haldar S, Jena N, Croce CM . 1995 Inactivation of Bcl-2 by phosphorylation Proc. Natl. Acad. Sci. USA 92: 4507–4511
Mizuno K, Kubo K, Saido TC, Akita Y, Osada S, Kuroki T, Ohno S, Suzuki K . 1991 Structure and properties of a ubiquitously expressed protein kinase C, nPKC delta Eur. J. Biochem. 202: 931–940
Denning MF, Dlugosz AA, Howett MK, Yuspa SH . 1993 Expression of an oncogenic rasHa gene in murine keratinocytes induces tyrosine phosphorylation and reduced activity of protein kinase C delta J. Biol. Chem. 268: 26079–26081
Geiges D, Marks F, Gschwendt M . 1995 Loss of protein kinase C delta from human HaCaT keratinocytes upon ras transfection is mediated by TGF alpha Exp. Cell Res. 219: 299–303
Takahashi T, Ogo M, Hibino T . 1998 Partial purification and characterization of two distinct types of caspases from human epidermis J. Invest. Dermatol. 111: 367–372
Boyce ST, Ham RG . 1983 Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture J. Invest. Dermatol. 81: 33s–40s
Mitra R, Nickoloff B . 1994 Cultivation of human epidermal keratinocytes in serum-free growth medium 17–19
Boukamp P, Petrussevska RT, Breitkreutz D, Homung J, Markham A, Fusenig NE . 1988 Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line J. Cell Biol. 106: 761–771
Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB . 1997 Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria Cell 91: 627–637
Kinsella TM, Nolan GP . 1996 Episomal vectors rapidly and stably produce high-titer recombinant retrovirus Hum. Gene Ther. 7: 1405–1413
Qin JZ, Chaturvedi V, Denning MF, Choubey D, Diaz MO, Nickoloff BJ . 1999 Role of NF-kappa B in the apoptotic-resistant phenotype of keratinocytes J. Biol. Chem. 274: 37957–37964
Acknowledgements
We thank all members of the Skin Cancer Research Program for their input into this project, especially Pat Bacon for help with cell culture, and Jeffrey Panella for construction of LZRS-Linker. We also thank Dr Elayne Bornslaeger (Northwestern University Medical School, Chicago, IL, USA) for help with construction of the FLAG epitope tagged vectors, Dr Ester Fernandez (National Institutes of Health, Bethesda, MD, USA) for help with mitochondria fractionation, and Linda Fox of the Loyola University Cell Imaging Core. We also thank Drs Paul Khavari and Garry P Nolan for providing us with the LZRS retroviral vector and Phoenix-Ampho packaging cells. We are grateful to Dr Norbert Fusenig for providing the HaCaT cell line. This work was supported in part by NIH grant CA83784 (MF Denning).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by B Osborne
Rights and permissions
About this article
Cite this article
Denning, M., Wang, Y., Tibudan, S. et al. Caspase activation and disruption of mitochondrial membrane potential during UV radiation-induced apoptosis of human keratinocytes requires activation of protein kinase C. Cell Death Differ 9, 40–52 (2002). https://doi.org/10.1038/sj.cdd.4400929
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400929
