
Abstract
When PC12 cells are deprived of trophic support they undergo apoptosis. We have previously shown that survival of trophic factor-deprived PC12M1 cells can be promoted by activation of the G protein-coupled muscarinic receptors. The mechanism whereby muscarinic receptors inhibit apoptosis is poorly understood. In the present study we investigated this mechanism by examining the effect of muscarinic receptor activation on the serum deprivation-induced activity of key players in apoptosis, the caspases, in PC12M1 cells. The results showed that m1 muscarinic activation inhibits caspase activity induced by serum deprivation. This effect appeared to be caused by the prevention of activation of caspases such as caspase-2 and caspase-3, and not by the inhibition of existing activity. Muscarinic receptor activation also stimulated the mitogen-activated protein kinase/extracellular signaling-regulated kinase (MAPK/ERK) and phosphoinositide (PI) 3-kinase signaling pathways. The PI 3-kinase pathway inhibitors wortmannin and LY294002, as well as the MAPK/ERK pathway PD98059 inhibitor, did not however suppress the inhibitory effect of the muscarinic receptors on caspase activity. The results therefore suggested that the muscarinic survival effect is mediated by a pathway that leads to caspase inhibition by MAPK/ERK- and PI 3-kinase-independent signaling cascades. Cell Death and Differentiation (2000) 7, 825–833
Similar content being viewed by others


Introduction
Apoptosis is a controlled cell death process which plays an important role during neuronal development and may underlie some neurodegenerative disorders.1,2,3 A key component of the apoptotic machinery is a family of proteases called caspases. These enzymes are first synthesized as inactive proenzymes which, in response to apoptotic stimuli, are cleaved to generate their active subunits.4 Most if not all cells depend on trophic factors for their survival and die apoptotically if deprived of their trophic support.5 The survival factors operate by activating their target receptors, which in turn transduce the survival signaling to the apoptotic machinery. Receptors that mediate the survival response include tyrosine kinase receptors for growth factors such as nerve growth factor (NGF)6,7 and insulin-like growth factor,8 as well as receptors for cytokines such as colony-stimulating factor and interleukin (IL)-29 IL-3,10 IL-4,11 IL-9.12 The nature of the intracellular signaling whereby these receptors inhibit apoptosis is now beginning to be unraveled. This is largely because of the identification of two signaling pathways, the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway8,13,14 and the phosphoinositide (PI) 3-kinase signaling pathway,15 as important components of the survival signaling process. How these second-messenger systems inhibit apoptosis is not fully understood, but the mechanisms appear to include inhibition of caspase activation and activity. It was shown, for example, that the downstream target of PI 3-kinase, the protein kinase B (PKB)/Akt,16 can phosphorylate and inhibit the activity of caspase-9,17 as well as of the pro-apoptotic member of the Bcl-2 family, Bad.18,19 PKB/Akt can however also be activated by PI 3-kinase independent mechanisms such as by protein kinase A (PKA)20 and calcium/calmodulin dependent kinase kinase21 and Bad can also be phosphorylated by MAPK/ERK, RSK22,23 and PKA24 pathways.
In addition to tyrosine kinase receptors and cytokine receptors, neuronal apoptosis can be inhibited by GTP-binding protein (G-protein)-coupled receptors such as the muscarinic acetylcholine receptors (mAChRs).25,26 The mechanism of action and the signaling pathways underlying this muscarinic inhibition are not known. The mAChRs comprise five subtypes, which can be divided into two groups according to their signaling mechanisms: m1, m3, and m5 mAChRs are preferentially coupled to the pertussis-insensitive Gq/G11 proteins that stimulate phosphoinositide hydrolysis, whereas m2 and m4 mAChRs are coupled to Gi/Go proteins that inhibit adenylyl cyclase.27,28 Activation of the m1, m3, m5 mAChR subgroups can induce activation of a large variety of signaling pathways, including the MAPK/ERK and the PI 3-kinase signaling cascade.29,30,31,32,33,34,35,36
The goal of the present study was to investigate the mechanisms whereby muscarinic receptors inhibit apoptosis and to determine whether it involves caspase inhibition. This was done using PC12M1 cells that were previously prepared in our laboratory. These cells express m1 muscarinic receptors, whose activation inhibits apoptosis induced by trophic factor deprivation.26 PC12 cells serve as a useful model system for studying neuronal apoptosis since the molecular details of this process have been relatively well characterized in these cells.6,37,38,39,40,41,42 It was shown, for example, that caspases are activated and needed for apoptosis of trophic factor-deprived PC12 cells.43,44,45,46,47,48 The results of this study showed that activation of the m1 muscarinic receptor inhibits caspase activation induced by serum deprivation in PC12M1 cells, suggesting that the muscarinic effect on survival might be mediated by this inhibitory process. Furthermore, muscarinic receptor activation stimulated the putative PI 3-kinase and MAPK/ERK survival pathways, but these pathways do not appear to mediate the muscarinic effect on caspase inhibition.
Results
Activation of m1 muscarinic receptors inhibits DEVDase activity
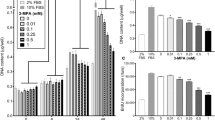
Apoptosis induced by trophic factor deprivation in PC12 cells requires caspase activity.43,46 We therefore examined the effect of muscarinic receptor activation on serum deprivation-induced caspase activity. Serum deprivation in PC12M1 cells resulted in rapid activation of caspases, as shown by cleavage of the peptide substrate DEVD-7-AMC (indicating DEVDase activity) in cell extracts prepared from the treated cells. This proteolytic activity was already evident 2 h after serum deprivation. It increased with time until it reached a peak about 3–6 h after serum deprivation, and then declined (Figure 1A). DEVDase activity at the plateau was about 10-fold higher (range 2–15-fold) than basal levels. Addition of the muscarinic agonist oxotremorine (100 μM) at the time of serum deprivation completely abolished this increase in DEVDase activity (Figure 1A). The inhibitory effect of oxotremorine on DEVDase was dose-dependent and saturable (Figure 1B), with a maximal effect at about 1 μM and an ED50 of approximately 0.05 μM. The oxotremorine-induced inhibition of DEVDase activity was blocked by the muscarinic antagonist atropine (Figure 1A), indicating that it was indeed muscarinically mediated.

Activation of muscarinic receptors block DEVDase activity induced by serum deprivation in PC12M1 cells. PC12M1 cells were deprived of serum and treated with 100 μM oxotremorine (oxo) or with 100 μM oxotremorine and 10 μM atropine (oxo+atr) for the indicated times (A). DEVDase activity was determined in lysates of the corresponding cells, as well as of untreated control serum-deprived cells, as described in Materials and Methods. The results shown are of a representative experiment (one of four experiments with similar results). Dose response of the effect of the muscarinic agonist oxotremorine (B) and m1-selective agonist AF102B (C) on DEVDase activity. Serum-deprived PC12M1 cells were treated with the indicated concentrations of oxotremorine or AF102B for 5 or 2.5 h respectively. DEVDase activity was determined for each treatment and in untreated control serum-deprived cells at t=0 and after 5 h. DEVDase activity (per cent of serum-deprived control cells) was determined by the measured ratio between DEVDase activity in agonist-treated cells, relative to the measured DEVDase activity in the absence of agonist. The data shown are mean values±S.D. (bars, n=3)
Although PC12M1 cells express both exogenous m1 and endogenous m4 receptor subtypes, the muscarinic survival effect is mediated by the m1 receptor.26 To determine whether the muscarinic-dependent inhibition of DEVDase activity is also mediated by m1 receptors, we examined the effect of AF102B, an m1-selective agonist,49 on the serum deprivation-induced caspase activity. As shown in Figure 1C, treatment with AF102B also inhibited the serum deprivation-induced DEVDase activity. The effect was dose-dependent and was maximal at about 10 μM with an ED50 of about 0.5 μM. Taken together, these results showed that activation of m1 muscarinic receptors inhibits caspase activity.
Muscarinic suppression of caspase activity is due to inhibition of caspase activation
In an attempt to characterize the muscarinic effect on DEVDase activity, we examined this effect when oxotremorine was added 2 or 4.5 h after serum deprivation. As shown in Figure 2, serum deprivation in PC12M1 cells resulted in a bell-shaped induction of DEVDase activity, which peaked 5 h after withdrawal of serum. Addition of oxotremorine 2 h after serum withdrawal, when DEVDase activity was rapidly increasing, stopped any further increase in DEVDase activity and kept it at a constant level for an additional 1.5 h. In contrast, addition of oxotremorine 4.5 h after serum withdrawal, i.e., at the peak of DEVDase activity and before its rapid decline, had no effect on DEVDase activity and was followed by a decline in the enzymatic activity similar to that of cells not treated with oxotremorine. These results suggested that muscarinic treatment prevents caspase activation, but does not affect the activity of caspases once they have been activated.

Muscarinic activation inhibits DEVDase activation but not DEVDase activity. PC12M1 cells were deprived of serum for the indicated times (♦). Oxotremorine (100 μM) was added to some of the cultures after 2 (○) or 4.5 h (▵) of serum deprivation. DEVDase activity was determined at the indicated time points and measured as described in Figure 1. The results shown are a representative experiment (one of two experiments with similar results)
DEVDase activity measures, inter alia, caspase-3 activity, and previous studies have shown that caspase-3 is activated during apoptosis induced by serum deprivation in PC12 cells.46 To investigate the mechanism underlying the muscarinic effect on caspases, we therefore examined the effect of oxotremorine on caspase-3 activation as indicated by the appearance of the large (p18) subunit of processed caspase-3 on immunoblot analysis. Serum deprivation in PC12M1 cells resulted in the appearance of the p18 processed subunit within 2 h; its levels increased over time, reaching a peak about 5 h after serum deprivation and then declining (data not shown). Treatment of serum-deprived PC12M1 cells with 100 μM oxotremorine prevented the formation of this caspase-3 subunit (Figure 3A). These results showed that muscarinic activation inhibits the processing of the inactive caspase-3 zymogen and the generation of its active subunits. Similar inhibition of caspase-3 processing was also observed when PC12M1 cells were treated with the survival factor NGF (Figure 3A). The finding that muscarinic activation inhibits caspase activation may suggest that the muscarinic survival effect is mediated via muscarinic inhibition of caspases. To further characterize the muscarinic effect on caspases, we attempted to determine whether muscarinic activation would inhibit the processing of caspase-2, a caspase shown to be directly related to apoptosis of serum-deprived PC12 cells.44,47 As shown in Figure 3B, in serum-deprived PC12M1 cells the 36–37-kDa N-terminal cleavage products of caspase-2 increased, indicating processing and activation of caspase-2. This processing was evident after 3 h and reached a peak about 5 h after serum deprivation. Treatment of serum-deprived PC12M1 cells with 100 μM oxotremorine inhibited the processing of caspase-2. Similar inhibition was observed when PC12M1 cells were treated with the survival factor NGF (Figure 3B), although NGF appeared to be slightly more effective than oxotremorine. These results suggested that muscarinic receptors may also inhibit activation of caspase-2.

Muscarinic activation inhibits the processing of pro-caspase-3 (A) and pro-caspase-2 (B). PC12M1-3 cells were deprived of serum and treated or untreated with 100 μM oxotremorine (O) or 50 ng/ml NGF (N) for the indicated times. Lysates of treated cells (100 μg protein) were resolved by SDS–PAGE on 12.5% polyacrylamide gels, and subjected to Western immunoblotting using caspase-3 or anti-N-Nedd2 antibody, as described in Materials and Methods. The results shown are of a representative experiment (one of three experiments with similar results)
We next examined the reversibility of the muscarinic effect on caspase activity. Serum-deprived PC12M1 cells were treated with oxotremorine and the muscarinic effect was then blocked by the addition of the muscarinic antagonist atropine 1, 2, 3, and 4 h after serum removal and oxotremorine treatment. As shown in Figure 4, blocking of muscarinic activation at these time points caused a time-dependent reduction in the effect of oxotremorine on DEVDase activity. These results suggested that the muscarinic effect on caspase activity is reversible and that continuous receptor activation is required for its full inhibitory effect.

Effects of transient muscarinic activation on DEVDase activity. PC12M1 cells were deprived of serum and treated or untreated with 100 μM oxotremorine (oxo) for 5 h. Atropine (10 μM) was added to the cultures 1, 2, 3, and 4 h after serum withdrawal and oxotremorine treatment. DEVDase activity was then determined 5 h after the start of the experiments. The results are expressed as described in Figure 1. The data shown are mean values±S.D. (bars, n=3)
Activation of muscarinic receptors in PC12M1 cells stimulates the MAPK/ERK and PI 3-kinase pathways, but these pathways are not needed for the muscarinic effect on caspases
Both the MAPK/ERK pathway and the PI 3-kinase pathway (via PKB/Akt) have been shown to play a role in the protective effects of various survival factors. To determine whether these signaling pathways can mediate the muscarinic effect on caspases in PC12M1 cells, we first investigated whether MAPK/ERK or PKB/Akt is stimulated by muscarinic activation. This was done by immunoblot experiments, using antibodies directed against the activated forms of these kinases (e.g., p-ERK and p-PKB/Akt). As shown in Figure 5, activation of the muscarinic receptors by oxotremorine induced phosphorylation of PKB/Akt as well as of ERK. Stimulation of PKB/Akt phosphorylation was already evident after 3 min (not shown), remained at about the same level for 11 h and then decreased (Figure 5A). Phosphorylation of PKB/Akt was blocked by preincubation of the cells with the potent PI 3-kinase inhibitors, wortmannin (100 nM) or LY294002 (10 μM) (Figure 5C), suggesting that the muscarinic activation in PC12M1 cells induces Akt/PKB phosphorylation by a PI 3-kinase-dependent pathway and that both wortmannin and LY294002 blocked this pathway in our experimental system. Treatment of the cells with oxotremorine also stimulated ERK phosphorylation (Figure 5B). This effect was already evident after 3 min, reached a peak after 10 min and then declined gradually until it became undetectable after 4 h. The muscarinic-dependent ERK phosphorylation was almost completely blocked by preincubation of the cells with the specific MAPK/ERK kinase inhibitor PD98059 (10 μM) (Figure 5D), suggesting that this muscarinic-dependent effect was mediated via the MAPK/ERK pathway and that, in our experimental system, PD98059 effectively blocked this pathway. These results suggested that activation of muscarinic receptors in PC12M1 cells stimulates both the MAPK/ERK and the PI 3-kinase pathways. The possibility that these pathways mediate the observed muscarinically induced inhibition of caspases was examined by measurement of the effect of the PI 3-kinase inhibitor wortmannin (100 nM) or PD98059 (100 μM) on the muscarinic effect on DEVDase activity. Incubation of serum-deprived cells with either wortmannin (Figure 6A) or PD98058 (Figure 6B) did not abolish the ability of oxotremorine to block caspase activity. Similar results were also obtained with higher concentrations of wortmannin (300 nM) and with the more stable inhibitor of PI 3-kinase, LY294002 (1–10 μM) (data not shown). Furthermore, wortmannin and PD98058 had no effect on the muscarinic inhibition of caspase activity when examined at other time points after serum deprivation (e.g., 5 and 2.5 h) or when added together and examined after 2.5 h (data not shown). Furthermore, neither wortmannin (100 nM), LY294002 (10 μM) nor PD98058 (10 μM) substantially reduced the ability of muscarinic activation to inhibit the processing of caspase-2 and caspase-3 (data not shown). Taken together, these results suggested that the MAPK/ERK and PI 3-kinase cascades are not needed for the muscarinic effect on caspase activity in PC12M1 cells.

Muscarinic activation stimulates the MAPK/ERK and PI 3-kinase/PKB/Akt pathways. PC12M1 cells were grown for about 18 h in the presence of 0.7% serum (A,B) or without serum (C,D) and 100 μM oxotremorine was then added to the cultures for the indicated times. Phosphorylation of PKB/Akt (P-Akt) (A,C) and of MAPK/ERK (P-ERK1 and P-ERK2) (B,D) was determined by immunoblot assays using antibodies directed against the phosphorylated forms of ERK1, ERK2 and PKB/Akt, as described in Materials and Methods. The levels of PKB/Akt or MAPK/ERK were determined by a similar method using antibodies directed against PKB/Akt or MAPK/ERK. For examining the effect of the inhibitors on PKB/Akt (C) or MAPK/ERK activation (D), PC12M1 cells were preincubated in the absence (-ser) or presence of 100 nM wortmannin (WT), 10 μM LY294002 (L) or 10 μM PD98059 for 30 min, and then with 100 μM oxotremorine (O) for an additional 10 min in the continued absence (-ser) or presence of these inhibitors. The data shown are representative experiments of four (A,B) or three (C), or two (D) independent experiments respectively, that yielded similar results

MAPK/ERK and PI 3-kinase pathways are not needed for the muscarinic effect on DEVDase activity. Serum-deprived PC12M1 cells were pretreated for 30 min in the absence (control) or presence of 100 nM wortmannin (WT) (A) or 100 μM PD98059 (B), and then incubated for an additional 2 h (without (control) or with wortmannin) or 4.5 h (without (control) or with PD98059) in the absence or presence of 100 μM oxotremorine (OXO). DEVDase activity was determined for each treatment and in control cells, as described in Figure 1. The empty squares correspond to DEVDase activity before the serum was removed. The filled squares correspond to DEVDase activity after serum withdrawal and the indicated pharmacological treatments. The results shown are a representative experiment (one of three experiments with similar results)
To determine whether these pathways mediate the muscarinically induced survival effect, we measured the extent to which the stable PI 3-kinase inhibitor LY294002 (10 μM) and the MAPK/ERK pathway inhibitor PD98059 (100 μM) influence the muscarinic effect on the viability of serum-deprived PC12M1 cells. Wortmannin was not used in these experiments as it is not stable in long-term treatments (e.g.,50). As shown in Figure 7, the addition of PD98059 to serum-deprived cells treated with oxotremorine did not significantly affect the viability of the cells, whereas addition of LY294002 or LY294002 plus PD980589 reduced their viability. However, the addition of these inhibitors by themselves could reduce cell viability. These results suggest that inhibition of the PI 3-kinase pathway in serum-deprived cells has a toxic effect on the cells, and that this toxic effect is countered by activation of the muscarinic receptors. However, the inability of oxotremorine treatment to completely block the death of serum-deprived cells in the presence of the inhibitors may suggest that the PI 3-kinase and the MAPK/ERK pathway have a partial role in the muscarinic survival effect.

Effect of LY294002 and PD98059 on muscarinic survival effect. PC12M1-3 cells were deprived of serum and then pretreated with 10 μM LY294002 or 100 μM PD98059 for 30 min prior to the addition of oxotremorine (OXO). The viability of the treated cells was determined, 24–28 h after serum deprivation, by the MTT assay, as described in Materials and Methods. Data are expressed as OD values in each treatment. The data shown are mean values±S.E. (bars, n=3)
Discussion
In this study we began to unravel the molecular pathways and processes whereby the G protein-coupled muscarinic receptors can inhibit apoptosis in neuronal cells. Our results showed that m1 muscarinic receptors inhibit apoptosis-induced caspase activation, but that the muscarinic pathway which mediates this effect is independent of the PI 3-kinase and MAPK/ERK activities.
Muscarinic signaling pathways
PI 3-kinase and its downstream target PKB/Akt, as well as MAPK/ERK, are known to deliver survival signals that inhibit apoptosis induced by withdrawal of growth factor in neurons and other types of cells. Our results suggest that these pathways are stimulated by activation of the muscarinic receptors. However, it cannot be concluded whether these pathways are essential for the muscarinic survival effect. The results show that the PI 3-kinase and MPAK/ERK pathway inhibitors partially inhibit the muscarinic survival effect, but it is not clear whether this inhibition is mediated by interference of the inhibitors with these specific pathways or by other mechanisms. Therefore our results do not prove that the PI 3-kinase and MAPK/ERK pathways are essential for the muscarinic survival effect in PC12M1 cells. We cannot exclude however the possibility that they participate in this effect. This assumption may be supported by a recent report which showed that activation of transiently transfected m1 receptors in COS-7 cells protects these cells against UV-induced apoptosis in a PI 3-kinase-dependent manner, probably through PKB/Akt.51
The finding of muscarinic-dependent stimulation of the PI 3-kinase and ERK pathways is in agreement with recent observations.51,52,53 It should be noted, however, that activation of PKB/Akt is not a general feature of all G protein-coupled receptors in PC12 cells, as the lysophosphatidic acid and thrombin receptors do not stimulate PKB/Akt in PC12 cells.54
Activation of muscarinic receptors inhibits caspase activation
In an attempt to understand the mechanism whereby muscarinic receptors inhibit apoptosis we directed our attention to one important aspect of this process, namely caspase activity. Caspase activity was determined by DEVDase activity as well as by the processing of caspase-2 and caspase-3. The DEVDase assay is widely used as an indicator of caspase activity and it measures the activities of caspases such as caspase-3 and -7.55,56,57 Serum deprivation for 3–6 h induced 2–15-fold increase in DEVDase activity. These differences in the induction of DEVDase activity are most likely due to fluctuation in the time required to reach the peak of activity between the different experiments. Accordingly, in some experiments, the same time point led to induction of DEVDase activity corresponding to the peak activity whereas in others it corresponded to the activity before or after the peak. Our results showed that activation of muscarinic receptors inhibits caspase activation induced by serum deprivation in PC12M1 cells. As caspases are needed for apoptosis induced by trophic factor deprivation in PC12 cells,43,46 the results shown suggested that the muscarinic survival effect is mediated at least in part by a pathway which leads to inhibition of caspases activity. Moreover, our finding that muscarinic activation inhibits pro-caspase-2 processing suggested that muscarinic activation inhibits caspase-2 activation. This further supports the notion that the muscarinic survival effect is mediated by inhibition of caspases, since caspase-2 plays an essential role in apoptosis of trophic factor-deprived PC12 cells.44,47 The role of other caspases, besides caspase-2 and caspase-3, in apoptosis induced by serum deprivation in PC12 cells has not yet been shown, although such a possibility cannot be excluded. The mechanism whereby muscarinic receptors inhibit caspase activation is presently unknown. A possible mechanism could be the inhibition cytochrome c release, either by increasing the expression of the anti-apoptotic members of the Bcl-2 family such as Bcl-2 and Bcl-x, or by other mechanism(s). It was recently shown that tyrosine kinase receptors, such as the NGF and epidermal growth factor receptors, can inhibit caspase activation.58,59 The findings presented here are the first demonstration that caspase activation can be inhibited by G protein-coupled receptors such as muscarinic receptors.
PC12M1 cells express both endogenous m4 and exogenous m1 receptors. We have previously shown that the muscarinic survival effect depends on m1 receptors.26 These findings were extended in this study by the observation that the m1-selective agonist AF102B is an effective inhibitor of caspase activity in trophic factor-deprived PC12M1 cells.
The signaling pathway mediating the muscarinic-dependent caspase inhibition
The results obtained showed that the PI 3-kinase and MAPK/ERK pathways are activated by muscarinic receptors in PC12M1 cells. These signaling pathways were previously shown in some systems to inhibit caspase activity.17,60 It was therefore of interest to examine their ability to transduce the muscarinic survival effect on caspases. Interestingly, none of the examined inhibitors of the PI 3-kinase or MAPK/ERK pathway could suppress the muscarinic effect on caspases, although all were able to inhibit the muscarinic-dependent phosphorylation of PKB/Akt or MAPK/ERK respectively. Furthermore, previous studies in PC12 cells have shown that similar concentrations of these inhibitors inhibit both survival effects and the activities of ERK and PI 3-kinase.8,15,61,62,63,64 Our results therefore suggested that the MAPK/ERK and PI 3-kinase pathways are not essential for mediating the muscarinic effect on caspase activity in PC12M1 cells, but rather that this effect is mediated by a new, as yet unidentified, pathway. This finding is in line with recent studies showing that in some cases the survival effects of growth factors and cytokine receptors are mediated by MAPK/ERK-independent and PI 3-kinase-independent pathways. For example, neither the survival effect of NGF on sympathetic neurons65 or Rat-1/MycER cells transfected with TrkA,66 nor that of granulocyte/macrophage colony-stimulating factor on MC/9 cells, is mediated by the PI 3-kinase pathway.67 Moreover, the MAPK/ERK and the PI 3-kinase pathways were not essential for the survival effect of NGF on apoptosis induced by ceramide in PC12 cells.64
One possible signaling pathway candidate for mediating the muscarinic effect on caspases might be sphingosine-1-phosphate, which was shown to play an important role in the survival effect of NGF on PC12 cells,68 and can be induced by at least the m2 and m3 muscarinic receptor subtypes.69 The finding that inhibition of the PI 3-kinase pathway partially attenuates the muscarinic survival effect on the viability of the cells but not on caspase inhibition, raises the question of the mechanism whereby these cells die under these conditions. One possible mechanism is that serum-deprived cells can die via both caspase-dependent and -independent pathways, as was shown in some apoptotic paradigms such as Bax-induced cell death in the presence of caspase inhibitors.70 Despite the fact that the caspase-dependent pathway seems to play a major role in the death of serum-deprived PC12 cells, once this pathway is inhibited, the caspase-independent pathway may take over. Alternatively, we cannot exclude the possibility that there are other caspases beside those examined in the present study which are activated and involved in apoptosis induced by trophic-factor-deprivation and that these caspases are inhibited by the muscarinic receptor in a different mechanism than that used to inhibit the DEVDase caspases and caspase-2. Accordingly it was shown that NGF withdrawal from differentiated PC12 cells induces expression of FasL which in turn may contribute to the apoptotic process via activation of the CD95 receptor71 In such a case it is still possible that the muscarinic receptors will inhibit the activation of caspase-8, the caspase which is directly activated when the CD95 is activated (for review see72), by a PI 3-kinase dependent mechanism as was shown for CD3 activation in Fas-treated Th2-type cells.73 In some systems, one signaling pathway appears to be sufficient for mediating survival induced by trophic agents such as NGF (PI 3-kinase15) and N-acetylcysteine (ERK63). In other systems, however, the survival effect may require the combined action of several signaling pathways. For example, insulin-like growth factor-1 inhibited apoptosis in differentiated PC12 cells by the combined action of PI 3-kinase and MAPK/ERK signaling pathways.8 The results of this study suggest that the muscarinic survival effect could be mediated by the combined effect of at least two different pathways. The major one leads to caspase inhibition and is independent of PI 3-kinase and ERK signaling. The other may involve the PI 3-kinase pathway, which leads to survival by a mechanism that does not affect caspases.
Materials and Methods
Materials
Ac-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Ac-DEVD-7AMC) was purchased from Biomol Research Laboratories (Plymouth Meeting, PA, USA). Wortmannin and LY294002 were purchased from Sigma (St. Louis, MO, USA). Each of the above reagents was resuspended as a 10 mM stock solution in DMSO. PD98059 was purchased from Promega (Madison, MI, USA) and resuspended as a 100 mM stock solution in DMSO. AF102B was obtained from Dr. A Fisher (Israel Institute for Biological Research, Israel) and resuspended as a 10 mM stock solution in water. NGF was purchased from Chemicon International (Harrow, UK). All other reagents were purchased from Sigma, unless otherwise stated.
Cell culture and treatment with various drugs
PC12M1 cells were established and grown as previously described.26,74 PC12M1-3, which was used in some experiments, is a subclone of PC12M1 cells and is highly sensitive to serum deprivation. For serum deprivation experiments, cultures were washed once with phosphate-buffered saline (PBS), detached (by 0.5 mM EDTA) and centrifuged. The cells were resuspended in PBS and centrifuged again. This resuspension and recentrifugation were repeated, and the resulting pellet was resuspended in RPMI 1640 medium and plated on rat tail collagen-coated plates. Oxotremorine and AF102B were added, unless otherwise indicated, at the time of serum deprivation. In the experiments aimed at examining the effects of various drugs on the muscarinic effects, PC12M1 cells were deprived of serum and maintained in the presence or absence of wortmannin, LY294002, PD98059, or atropine for 30 min. Oxotremorine was then added for the indicated times in the continued presence of the drugs.
Assay for DEVDase activity
Caspase activity was measured in terms of assayed DEVDase activity. PC12M1 cells were deprived of serum for different periods of time. At the end of each time period, cells (5×106) were collected, centrifuged, washed with PBS, and frozen at −70°C. After collection the pelleted cells were resuspended in 100 μl of extraction buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA and 10 mM EGTA) and lysed by three rounds of freezing and thawing. The extracts were then centrifuged for 5 min at 20 800×g. The resulting supernatants were normalized for protein content (approximately 300 μg) and assayed for DEVDase activity using the fluorescent synthetic peptide Ac-DEVD-7AMC (50 μM) in the reaction buffer (50 mM Tris-HCl, pH 7.4, 1 mM DTT and 2 mM MgCl2). Fluorescence at 360 nm for excitation and at 460 nm emission was measured after incubation for 30 min at 37°C. In each experiment, DEVDase activity was determined at t=0 and at the indicated times after serum deprivation in the absence or presence of oxotremorine and/or the indicated reagents. DEVDase activity induced by serum deprivation in the absence of oxotremorine was designated as the measured activity at the indicated time, minus the DEVDase activity at t=0.
Immunoblotting
For ERK and PKB/Akt immunoblotting, PC12M1 cells were grown for about 18 h in the presence of 0.7% serum or in its absence and then treated for 30 min with either wortmannin or PD98059. Oxotremorine was then added for an additional 3 min to 30 h. Cell lysates were prepared by incubation of the cells for 20 min on ice in lysis buffer (50 mM HEPES, pH 7.5, 10% glycerol, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 1.5 mM MgCl2, 200 μM sodium vanadate and protease inhibitor cocktail diluted 1 : 100 (Calbiochem Cat. No. 539131)) followed by centrifugation for 10 min at 20 800×g. For caspase-3 and caspase-2 immunoblotting, cell lysates were prepared from PC12M1-3 cells which had been deprived of serum for 2, 3, or 5 h, by three rounds of freezing and thawing in 100 μl of extraction buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA and 10 mM EGTA) followed by centrifugation for 5 min at 20 800×g. Aliquots of the resulting supernatants were subjected to sodium dodceyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) (10% polyacrylamide for ERK and PKB/Akt or 12.5% polyacrylamide for caspase-2 and caspase-3) and electroblotted onto supported nitrocellulose. Equal amounts of protein (80–100 μg) were loaded in each lane. Uniformity of sample loading was verified by staining of the blot with Ponceau. Blots were blocked for 1 h in 50 mM Tris base, pH 7.6, 150 mM NaCl, 0.1% Tween 20 containing 5% fat-free milk or in 10 mM Tris base, pH 7.6, 150 mM NaCl, 0.05% Tween 20 containing 5% fat-free milk for ERK or PKB/Akt and caspase-3, respectively. They were then incubated for 16 h with the primary antibody, rabbit polyclonal anti caspase-3 (CPP32) (1 : 2000) (Santa Cruz) or mouse anti-MAPK activated (diphosphorylated ERK-1&2) (1 : 15 000) (Sigma) or rabbit polyclonal anti-MAPK (1 : 5000) (Santa Cruz) or rabbit polyclonal anti Akt (1 : 2000) (New England Biolabs, Beverley, MA, USA) or rabbit anti phosphorylated Akt (phosph-Akt (Ser473) (1 : 2000) (New England Biolabs, Beverley, MA, USA)). Goat anti rabbit (1 : 10 000) or goat anti mouse IgG peroxidase conjugate (1 : 7500) was used as second antibody. For caspase-2 immunoblotting, blots were treated with anti-N-Nedd, an antibody directed against the N terminus of caspase-2, as previously described.44,58,75 Blots were developed using the Amersham Enhanced Chemiluminescence Kit.
Counting of viable cells
The number of living cells in 96-well plates (2×104 cells per well) was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.76 MTT was dissolved in PBS at a concentration of 5 mg/ml. From this stock solution, 10 μl per 100 μl of medium were added to each well, and the plates were incubated at 37°C for 3 h. Acid-isopropanol (100 μl of 0.04 M HCl in isopropanol) was then added to the well and mixed in. After 15 min at room temperature, the plates were read on a Micro-ELISA reader at a test wavelength of 550 nm and a reference wavelength of 650 nm.
Abbreviations
- Ac-DEVD-7AMC:
-
Ac-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin
- ERK:
-
extracellular signal-regulated kinase
- G-protein:
-
GTP-binding protein
- MAPK:
-
mitogen-activated protein kinase
- mAChRs:
-
muscarinic acetylcholine receptors
- MTT:
-
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
- NGF:
-
nerve growth factor
- PBS:
-
phosphate-buffered saline
- PI:
-
phosphoinositide
- PKB:
-
protein kinase B
References
Barinaga M . (1998) Is apoptosis key in Alzheimer's disease? Science 281: 1303–1304
Barinaga M . (1998) Stroke-damaged neurons may commit cellular suicide. Science 281: 1302–1303
Bergeron L and Yuan J . (1998) Sealing one's fate: control of cell death in neurons. Curr. Opin. Neurobiol. 8: 55–63
Thornberry NA and Lazebnik Y . (1998) Caspases: enemies within. Science 281: 1312–1316
Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y and Jacobson MD . (1994) Programmed cell death and the control of cell survival. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 345: 265–268
Rukenstein A, Rydel RE and Green LA . (1991) Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J. Neurosci. 11: 2552–2563
Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA and Barbacid M . (1994) Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 368: 246–249
Parrizas M, Saltiel AR and LeRoith D . (1997) Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J. Biol. Chem. 272: 154–161
Armant M, Delespesse G and Sarfati M . (1995) IL-2 and IL-7 but not IL-12 protect natural killer cells from death by apoptosis and up-regulate bcl-2 expression. Immunology 85: 331–337
Williams GT, Smith CA, Spooncer E, Dexter TM and Taylor DR . (1990) Haemopoietic colony stimulating factors promote cell survival by suppressing apoptosis. Nature 343: 76–79
Vella A, Teague TK, Ihle J, Kappler J and Marrack P . (1997) Interleukin 4 (IL-4) or IL-7 prevents the death of resting T cells: stat6 is probably not required for the effect of IL-4. J. Exp. Med. 186: 325–330
Demoulin JB, Uyttenhove C, Van Roost E, DeLestre B, Donckers D, Van Snick J and Renauld JC . (1996) A single tyrosine of the interleukin-9 (IL-9) receptor is required for STAT activation, antiapoptotic activity, and growth regulation by IL-9. Mol. Cell. Biol. 16: 4710–1716
Xia Z, Dickens M, Raingeaud J, Davis RJ and Greenberg ME . (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326–1331
Anderson CNG and Tolkovsky AM . (1999) A role for MAPK/ERK in sympathetic neuron survival: protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J. Neurosci. 19: 664–673
Yao R and Cooper GM . (1995) Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 267: 2003–2006
Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR and Greenberg ME . (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661–665
Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S and Reed JC . (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282: 1318–1321
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y and Greenberg ME . (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241
del Peso L, Gonzalez-Garcia M, Page C, Herrara R and Nunez G . (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278: 687–689
Filippa N, Sable CL, Filloux C, Hemmings B and Van Obberghen E . (1999) Mechanism of protein kinase B activation by cyclic AMP-dependent protein kinase. Mol. Cell. Biol. 19: 4989–5000
Yano S, Tokumitsu H and Soderling TR . (1998) Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396: 584–587
Scheid MP, Schubert KM and Duronio V . (1999) Regulation of bad phosphorylation and association with Bcl-x(L) by the MAPK/Erk kinase. J. Biol. Chem. 274: 31108–31113
Bonni A, Brunet A, West AE, Datta SR, Takasu MA and Greenberg ME . (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms [see comments]. Science 286: 1358–1362
Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD and Korsmeyer SJ . (1999) Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell 3: 413–422
Yan GM, Lin SZ, Irwin RP and Paul SM . (1995) Activation of muscarinic cholinergic receptors blocks apoptosis of cultured cerebellar granule neurons. Mol. Pharmacol. 47: 248–257
Lindenboim L, Pinkas-Kramarski R, Sokolovsky M and Stein R . (1995) Activation of muscarinic receptors inhibits apoptosis in PC12M1 cells. J. Neurochem. 64: 2491–2499
Wess J . (1993) Molecular basis of muscarinic acetylcholine receptor function. Trends Pharmacol. Sci. 14: 308–313
Wess J . (1996) Molecular biology of muscarinic acetylcholine receptors. Crit. Rev. Neurobiol. 10: 69–99
Fukuda K, Higashida H, Kubo T, Maeda A, Akiba I, Bujo H, Mishina M and Numa S . (1988) Selective coupling with K+ currents of muscarinic acetylcholine receptor subtypes in NG108-15 cells. Nature 335: 355–358
Fukuda K, Kubo T, Akiba I, Maeda A, Mishina M and Numa S . (1987) Molecular distinction between muscarinic acetylcholine receptor subtypes. Nature 327: 623–625
Felder CC, Poulter MO and Wess J . (1992) Muscarinic receptor-operated Ca2+ influx in transfected fibroblast cells is independent of inositol phosphates and release of intracellular Ca2+. Proc. Natl. Acad. Sci. U.S.A. 89: 509–513
Sandmann J, Peralta EG and Wurtman RJ . (1991) Coupling of transfected muscarinic acetylcholine receptor subtypes to phospholipase D. J. Biol. Chem. 266: 6031–6034
Marais R, Light Y, Mason C, Paterson H, Olson MF and Marshall CJ . (1998) Requirement of Ras-GTP-Raf complexes for activation of Raf-1 by protein kinase C. Science 280: 109–112
Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS and Wetzker R . (1997) Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science 275: 394–397
van Biesen T, Hawes BE, Raymond JR, Luttrell LM, Koch WJ and Lefkowitz RJ . (1996) G(o)-protein alpha-subunits activate mitogen-activated protein kinase via a novel protein kinase C-dependent mechanism. J. Biol. Chem. 271: 1266–1269
Schmidt M, Voss M, Thiel M, Bauer B, Grannass A, Tapp E, Cool RH, de Gunzburg J, von Eichel-Streiber C and Jakobs KH . (1998) Specific inhibition of phorbol ester-stimulated phospholipase D by Clostridium sordellii lethal toxin and Clostridium difficile toxin B-1470 in HEK-293 cells. Restoration by Ral GTPases. J. Biol. Chem. 273: 7413–7422
Lindenboim L, Diamond R, Rothenberg E and Stein R . (1995) Apoptosis induced by serum deprivation of PC12 cells is not preceded by growth arrest and can occur at each phase of the cell cycle. Cancer Res. 55: 1242–1247
Park DS, Stefanis L, Yan CYL, Farinelli SE and Greene LA . (1996) Ordering the cell death pathway. Differential effects of BCL2, an interleukin-1-converting enzyme family protease inhibitor, and other survival agents on JNK activation in serum/nerve growth factor-deprived PC12 cells. J. Biol. Chem. 271: 21898–21905
Ochu EE, Rothwell NJ and Waters CM . (1998) Caspases mediate 6-hydroxydopamine-induced apoptosis but not necrosis in PC12 cells. J. Neurochem. 70: 2637–2640
Suzuki A . (1997) Amyloid beta-protein induces necrotic cell death mediated by ICE cascade in PC12 cells. Exp. Cell Res. 234: 507–511
Yen C, Mar M and Zeisel SH . (1999) Choline deficiency-induced apoptosis in PC12 cells is associated with diminished membrane phosphatidylcholine and sphingomyelin, accumulation of ceramide and diacylglycerol, and activation of a caspase. FASEB J. 13: 135–142
Kruman I, Guo Q and Mattson MP . (1998) Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J. Neurosci. Res. 51: 293–308
Troy CM, Stefanis L, Prochiantz A, Greene LA and Shelanski ML . (1996) The contrasting roles of ICE family proteases and interleukin-1beta in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc. Natl. Acad. Sci. U.S.A. 93: 5635–5640
Troy CM, Stefanis L, Greene LA and Shelanski ML . (1997) Nedd2 is required for apoptosis after trophic factor withdrawal, but not superoxide dismutase (SOD1) downregulation, in sympathetic neurons and PC12 cells. J. Neurosci. 17: 1911–1918
Stefanis L, Park DS, Yan CY, Farinelli SE, Troy CM, Shelanski ML and Greene LA . (1996) Induction of CPP32-like activity in PC12 cells by withdrawal of trophic support. Dissociation from apoptosis. J. Biol. Chem. 271: 30663–30671
Haviv R, Lindenboim L, Li H, Yuan J and Stein R . (1997) Need for caspases in apoptosis of trophic factor-deprived PC12 cells. J. Neurosci. Res. 50: 69–80
Haviv R, Lindenboim L, Yuan J and Stein R . (1998) Need for caspase-2 in apoptosis of growth-factor-deprived PC12 cells. J. Neurosci. Res. 52: 491–497
Schulz JB, Bremen D, Reed JC, Lommatzsch J, Takayama S, Wullner U, Loschmann PA, Klockgether T and Weller M . (1997) Cooperative interception of neuronal apoptosis by BCL-2 and BAG-1 expression: prevention of caspase activation and reduced production of reactive oxygen species. J. Neurochem. 69: 2075–2086
Gurwitz D, Haring R, Pinkas Kramarski R, Stein R, Heldman E, Karton Y and Fisher A . (1995) NGF-dependent neurotrophic-like effects of AF102B, an M1 muscarinic agonist, in PC12M1 cells. Neuroreport 6: 485–488
Kimura K, Hattori S, Kabuyama Y, Shizawa Y, Takayanagi J, Nakamura S, Toki S, Matsuda Y, Onodera K and Fukui Y . (1994) Neurite outgrowth of PC12 cells is suppressed by wortmannin, a specific inhibitor of phosphatidylinositol 3-kinase. J. Biol. Chem. 269: 18961–18967
Murga C, Laguinge L, Wetzker R, Cuadrado A and Gutkind JS . (1998) Activation of Akt/protein kinase B by G protein-coupled receptors. A role for alpha and beta gamma subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinasegamma. J. Biol. Chem. 273: 19080–19085
Haring R, Fisher A, Marciano D, Pittel Z, Kloog Y, Zuckerman A, Eshhar N and Heldman E . (1998) Mitogen-activated protein kinase-dependent and protein kinase C-dependent pathways link the m1 muscarinic receptor to beta-amyloid precursor protein secretion. J. Neurochem. 71: 2094–2103
Desdouits-Magnen J, Desdouits F, Takeda S, Syu LJ, Saltiel AR, Buxbaum JD, Czernik AJ, Nairn AC and Greengard P . (1998) Regulation of secretion of Alzheimer amyloid precursor protein by the mitogen-activated protein kinase cascade. J. Neurochem. 70: 524–530
Andjelkovic M, Suidan HS, Meier R, Frech M, Alessi DR and Hemmings BA . (1998) Nerve growth factor promotes activation of the alpha, beta and gamma isoforms of protein kinase B in PC12 pheochromocytoma cells. Eur. J. Biochem. 251: 195–200
Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD and Wong WW . (1997) Substrate specificities of caspase family proteases. J. Biol. Chem. 272: 9677–9682
Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT and Nicholson DW . (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272: 17907–17911
Janicke RU, Ng P, Sprengart ML and Porter AG . (1998) Caspase-3 is required for alpha-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J. Biol. Chem. 273: 15540–15545
Stefanis L, Troy CM, Qi H, Shelanski ML and Greene LA . (1998) Caspase-2 (Nedd-2) processing and death of trophic factor-deprived PC12 cells and sympathetic neurons occur independently of caspase-3 (CPP32)-like activity. J. Neurosci. 18: 9204–9215
Lan L and Wong NS . (1999) Phosphatidylinositol 3-kinase and protein kinase C are required for the inhibition of caspase activity by epidermal growth factor. FEBS Lett. 444: 90–96
Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN and Hay N . (1997) The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 11: 701–713
Spear N, Estevez AG, Barbeito L, Beckman JS and Johnson GV . (1997) Nerve growth factor protects PC12 cells against peroxynitrite-induced apoptosis via a mechanism dependent on phosphatidylinositol 3-kinase. J. Neurochem. 69: 53–59
Kummer JL, Rao PK and Heidenreich KA . (1997) Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J. Biol. Chem. 272: 20490–20494
Yan CYI and Greene LA . (1998) Prevention of PC12 cell death by N-acetylcysteine requires activation of the Ras pathway. J. Neurosci. 18: 4042–4049
Hartfield PJ, Bilney AJ and Murray AW . (1998) Neurotrophic factors prevent ceramide-induced apoptosis downstream of c-Jun N-terminal kinase activation in PC12 cells. J. Neurochem. 71: 161–169
Philpott KL, McCarthy MJ, Klippel A and Rubin LL . (1997) Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J. Cell Biol. 139: 809–815
Ulrich E, Duwel A, Kauffmann-Zeh A, Gilbert C, Lyon D, Rudkin B, Evan G and Martin-Zanca D . (1998) Specific TrkA survival signals interfere with different apoptotic pathways. Oncogene 16: 825–832
Scheid MP and Duronio V . (1998) Dissociation of cytokine-induced phosphorylation of Bad and activation of PKB/akt: involvement of MEK upstream of Bad phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 95: 7439–7444
Edsall LC, Pirianov GG and Spiegel S . (1997) Involvement of sphingosine 1-phosphate in nerve growth factor-mediated neuronal survival and differentiation. J. Neurosci. 17: 6952–6960
Meyer zu Heringdorf D, Lass H, Alemany R, Laser KT, Neumann E, Zhang C, Schmidt M, Rauen U, Jakobs KH and van Koppen CJ . (1998) Sphingosine kinase-mediated Ca2+ signalling by G-protein-coupled receptors. EMBO J. 17: 2830–2837
Xiang J, Chao DT and Korsmeyer SJ . (1996) BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc. Natl. Acad. Sci. U.S.A. 93: 14559–14563
Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR and Karin M . (1999) Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to fas ligand induction and cell death. Mol. Cell. Biol. 19: 751–763
Ashkenazi A and Dixit VM . (1998) Death receptors: signaling and modulation. Science 281: 1305–1308
Varadhachary AS, Peter ME, Perdow SN, Krammer PH and Salgame P . (1999) Selective up-regulation of phosphatidylinositol 3′-kinase activity in Th2 cells inhibits caspase-8 cleavage at the death-inducing complex: a mechanism for Th2 resistance from Fas-mediated apoptosis. J. Immunol. 163: 4772–4779
Pinkas Kramarski R, Stein R, Lindenboim L and Sokolovsky M . (1992) Growth factor-like effects mediated by muscarinic receptors in PC12M1 cells. J. Neurochem. 59: 2158–2166
Stefanis L, Troy CM, Qi H and Greene LA . (1997) Inhibitors of trypsin-like serine proteases inhibit processing of the caspase Nedd-2 and protect PC12 cells and sympathetic neurons from death evoked by withdrawal of trophic support. J. Neurochem. 69: 1425–1437
Mosmann T . (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65: 55–63
Acknowledgements
This work was supported by the German-Israeli Foundation for Scientific Research and Development and the Joint German-Israeli Research Program. We thank Dr. Lloyd Greene for providing the anti-N-Nedd antibody and Ms. Shirley Smith for excellent editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by C Borner
Rights and permissions
About this article
Cite this article
Leloup, C., Michaelson, D., Fisher, A. et al. M1 muscarinic receptors block caspase activation by phosphoinositide 3-kinase- and MAPK/ERK-independent pathways. Cell Death Differ 7, 825–833 (2000). https://doi.org/10.1038/sj.cdd.4400713
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400713
Keywords
This article is cited by
-
M1 Muscarinic Receptor Deficiency Attenuates Azoxymethane-Induced Chronic Liver Injury in Mice
Scientific Reports (2015)
-
M1 muscarinic acetylcholine receptor in Alzheimer’s disease
Neuroscience Bulletin (2014)
-
Cholinergic receptor pathways involved in apoptosis, cell proliferation and neuronal differentiation
Cell Communication and Signaling (2009)
-
Effect of Novel Amino Acids and Dipeptides Substituted 3-Morpholino Arecoline Derivatives as Muscarinic Receptor 1 Agonists in Alzheimer’s Dementia Models
International Journal of Peptide Research and Therapeutics (2009)
-
Functional Characterization of the Epidermal Cholinergic System In Vitro
Journal of Investigative Dermatology (2006)
