
Abstract
A minority of centroblastic and centroblastic/centrocytic cell lymphomas are accompanied by a prominent epithelioid cell response and were suggested to be a distinct variant of B-cell lymphoma of germinal center cell origin. To confirm the clinicopathologic significance of these mainly large B-cell lymphomas with an epithelioid cell response (LBCL-ER), we reviewed 50 patients with LBCL-ER and compared the results with those of 167 other diffuse large B-cell lymphomas (DLBCL) and 94 follicular lymphomas (FL) without epithelioid response. The patients with LBCL-ER showed a higher age distribution (median 71, P = .03), a female predominance (M:F = 18:32, P = .001) and less frequent involvement of extranodal sites >1 (P = .004) compared with those with DLBCL, and presented with a bulky mass of the affected lymph nodes in 54% of cases. They were also older (P = .0006) and more associated with the aggressive clinical factors such as serum LDH level and International Prognostic Index score than those with FL. Histologically, nine cases (18%) partially showed a follicular growth pattern, and the others (82%) were occupied by a diffuse growth pattern. The epithelioid cells were accumulated in large demarcated masses, partially imparting a lymphoepithelioid (Lennert) lymphoma-like appearance to some portions of the lesions in every case. Immunohistochemically, LBCR-ER was positive for CD20 in every case, CD10 in 43% of the cases, and BCL-2 in 56%. None of the tumor cells in the 40 cases tested expressed CD5 antigen. Immunostaining also often highlighted the remnants of the follicular dendritic cell network. The BCL-2 gene rearrangement was detected in only 19% of the cases examined. The survival curve of the cases of LBCL-ER was almost identical with that of DLBCL and was significantly inferior to that of FL. The centroblastic and centroblastic/centrocytic lymphoma with an epithelioid cell response may be regarded as the morphologic variant of DLBCL preferentially arising in the aged population and reflecting the disease progression of FL.
Similar content being viewed by others

INTRODUCTION
Diffuse large B-cell lymphoma is a heterogeneous category of lymphoma awaiting reproducible criteria to define meaningful subtypes (1, 2, 3, 4). The lymphomatous infiltrate of large B-cell lymphoma is sometimes accompanied by a reactive component comprising small lymphocytes, plasma cells, eosinophils, and histiocytes, which may be a landmark for identifying some distinct variants or entities and represent their biological behavior (2, 4). T-cell/histiocyte-rich B-cell lymphoma (TCRBCL) is now regarded as a rare variant of large B-cell lymphoma and features a prominent component of reactive T lymphocytes with various numbers of histiocytes, which may have an epithelioid appearance (5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20). In recent years, a few studies have considered TCRBCL to be synonymous with CD20-positive Hodgkin’s disease or an aggressive variant of nodular lymphocyte predominant Hodgkin’s disease (8, 9, 15, 16). These cases are different from earlier cases reported as TCRBCL, which is a heterogeneous disease encompassing follicular center cell lymphoma and non-follicular center large B-cell lymphoma (5, 6, 10), although some areas of disagreement and controversy surrounding the diagnosis of TCRBCL remain (12, 17, 18, 20).
Lymphoplasmacytoid immunocytoma in the Kiel classification is also often accompanied by an epithelioid response and is currently regarded as marginal zone B-cell lymphoma of a mucosa-associated lymphoid tissue (MALT) type at an extranodal site (2, 3, 21). We previously reported 12 cases of centroblastic and centroblastic/centrocytic lymphomas associated with a prominent epithelioid cell response (22). These mainly large B-cell lymphomas with an epithelioid cell response (LBCL-ER) are characterized by clinical presentation as a bulky mass, spanning between diffuse large B-cell lymphoma and follicular lymphoma on morphology, and have been suggested to be of germinal center cell origin in many cases. For these reasons, we asserted that LBCL-ER are distinct from TCRBCL and immunocytoma and may merit separate consideration because of the problem posed by the initial diagnosis and therapeutic approach. However, the clinical significance of this tumor is still unconfirmed. This prompted us to investigate the clinicopathologic and immunohistochemical features of LBCL-ER in a large patient population. To confirm the clinicopathological significance of LBCL-ER, we performed a retrospective study on 50 cases with such tumors, and compared the results with those of 167 diffuse large B-cell lymphomas and 94 follicular lymphomas without epithelioid cell response.
MATERIALS AND METHODS
Patient Selection
A total of 50 cases of LBCL-ER were collected, 24 from the series of one of the authors (MK) and 26 from Aichi Cancer Center Hospital (ACCH), 14 of which accounted for 2.86% of the 489 cases consecutively diagnosed as diffuse centroblastic lymphoma at ACCH between 1979 and 1999. Eleven cases (Cases 4–6, 9, 10, 14, 15, 29, 30, 34, and 49) had been reported previously (22).
Clinical information was obtained from the hospital records and/or the attending physicians. The extent of disease was determined according to the Ann Arbor system (23
In 49 of the cases, lymph node biopsy specimens obtained at presentation were available, and a tonsillar specimen was obtained from the other case (Case 26). In two cases (Cases 22 and 25), bone marrow aspiration specimens were also available. In four patients, biopsy specimens of recurrent lesions were also obtained: lymph nodes from three cases (Cases 14, 20, and 24) and a tonsil from one (Case 35).
For the control group, 167 patients with diffuse large B-cell lymphoma without an epithelioid cell response (DLBCLFP−) and follicular large cell lymphoma without an epithelioid cell response (FLCLEP−). They were diagnosed consecutively and received treatment between 1983 and 2000 at ACCH.
Histopathology
Formalin-fixed paraffin-embedded specimens were prepared, and 3-micrometer sections from them were cut and stained with hematoxylin and eosin (HE), by the periodic acid—Schiff method, with Giemsa stain, and by silver impregnation (Watanabe). The 50 cases were selected by the following criteria: (1) at low magnification, epithelioid granulomatous response occupied 75% of the lymph node (Fig. 1A) and (2) the granulomatous lesion usually contained >50 epithelioid cells per 400 × microscopic field.

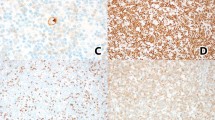
A, low-power field of an affected node. Geographic epithelioid granulomatous response occupied >90% of the lymph node. (Case 5). Hematoxylin and eosin (HE), 10 ×. B, low-power field of an affected lymph node. Tubercular fibrosis compartmentalized the parenchyma producing a vague nodular pattern. Note the epithelioid cell accumulation in large and poorly demarcated masses (Case 5). HE 10 ×. C, high-power field of the affected lymph node. The tumor cells had large round or indented nuclei with small- to medium-sized nucleoli (Case 13). HE 100 ×. D, high-power field of the affected lymph node. Note the binucleated cells. Epithelioid cells are diffusely infiltrating. (Case 24). HE 100 ×. E, positive membrane staining of tumor cells with CD10 antigens (Case 30). SAB method 100 ×. F, positive membrane staining of mononuclear cells and spindle-shaped cells with CAN.42 antibody (Case 13). SAB method; magnification, 100 ×.
Immunophenotypic Study
In an immunohistochemical study, sections were stained by the streptavidin-biotin-peroxidase (SAB) method (ScyTec, Logan, UT or Immunotech, Marseille, France) using antibodies against human immunoglobulin light chains (MBL Co., Nagoya, Japan or DAKO A/S, Glostrup, Denmark), 4C7 (CD5; Novocastra, Newcastle, UK), 56C6 (CD10; Novocastra), L26 (CD20; Nichirei Co., Tokyo, Japan), 1F8 (CD21; Dako), 1B12 (CD23; Novocastra), UCHL-1 (CD45RO; Nichirei Co), KP-1 (CD68; DAKO), anti-follicular dendritic reticulm cell (FDC) antibody (CNA.42; DAKO), and BCL-2 protein (DAKO).
When necessary, Leu M1 (CD15; Immunotech, Marseille, France), Ber-H2 (CD30; DAKO), and/or EMA (Novocastra) were also stained by the SAB method. Microwave antigen retrieval was used with 4C7, 56C6, 1F8, 1B12, BerH-2, CNA.42, and BCL-2 immunostains.
In Situ Hybridization Study
In situ hybridization (ISH) with Epstein Barr virus (EBV)-encoded small RNA (EBER) oligonucleotides was used to test for the presence of EBV small RNAs in formalin-fixed paraffin-embedded sections according to the manufacturer’s instructions (Novocastra).
Genotypic Study
DNA was extracted from formalin-fixed paraffin-embedded tissues in selected cases. The purified DNA was used to analyze the presence of the bcl-2/IgH translocation at the major breakpoint region. This study was performed by use of a polymerase chain reaction (PCR) technique described previously (24
Statistical Analysis
Correlations between the two groups were examined with the chi-square test, the Fisher exact test, Student’s t test, and the Mann-Whitney U test. Patient survival data were analyzed with the Kaplan-Mayer method and were compared by means of the log-rank test. Univariable analysis was performed with the Cox proportional hazard regression model. Data were analyzed with the SAS system software (SAS Institute Inc., Cary, NC).
RESULTS
Clinicopathological, immunohistochemical, and genotypic findings are shown in Tables 1 through 3
Histological Findings
All of the specimens in this study exhibited the histological features of centroblastic (n = 46) or centroblastic/centrocytic lymphomas (n = 4) with a prominent epithelioid response (22). Nine lesions (18%; Cases 6, 15–17, 20, 24, 26, 30, and 35) displayed a partial follicular growth pattern, and the other 41 (82%) showed a diffuse growth pattern. At low magnification, thick trabecular fibrosis compartmentalized the lymph node parenchyma in 39 cases, producing a vague nodular pattern even in the diffuse lymphomas (Fig. 1B). The tumors occasionally contained areas of monotonous proliferation of lymphoma cells without a significant epithelioid granulomatous response. These tumor cells were also difficult to recognize against the background of prominent epithelioid cell infiltration in other areas of the lymph node lesions. The majority of the tumor cells had round nuclei with fine nuclear chromatin and two to four small nucleoli, which were frequently situated at the nuclear membrane (Fig. 1C). A small number of immunoblasts, immunoblast-like cells, and medium-sized cells with cleaved nuclei were also seen (Fig. 1C). A few multilobulated centroblasts and multinucleated cells somewhat resembling Reed-Sternberg cells were observed in two cases (Cases 24 and 38; Fig. 1D). At low power field, the epithelioid cells were usually accumulated in large and poorly demarcated masses (Fig. 1b). A few Langerhans’ giant cells were observed in two cases (Cases 20 and 38). No reactive cells other than epithelioid cells were prominent. Scattered tingible body macrophages were intermingled with tumor cells in one case (Case 2), and focal lymph node necrosis was also seen in only one case (Case 38). In one case each, there was a preceding episode of follicular lymphoma (Case 1) and diffuse large B-cell lymphoma (Case 45) without an epithelioid cell response. The cellular infiltrates in the bone marrow of two patients (Cases 22 and 25) were virtually identical to those in the corresponding nodal sections.
Recurrent lesions were examined in four cases (Cases 14, 20, 24, and 35). Macroaggregated epithelioid cells had persisted in one case (Case 14), but the epithelioid cell response had resolved in the other three cases (Cases 20, 24, and 35).
Immunohistochemical Findings
The tumor cells in all cases were positive for CD20, but not for CD3, CD45RO, or CD68. Monoclonal intracytoplasmic immunoglobulin was detected in five of the 32 cases tested. The tumor cells did not express CD5 antigen in any of the 40 cases examined thus far. The majority of the neoplastic cells expressed CD10 antigen in 18 cases (43%), including five follicular lymphomas, among the 42 cases tested (Fig. 1D). The two specimens containing multilobulated centroblasts and Reed-Sternberg-like tumor cells (Cases 24 and 38) were also CD10 positive. Some of the multilobulated centroblasts and Reed-Sternberg like tumor cells in one (Case 38) of the two specimens expressed CD30 antigen, but they were negative for CD15 and EMA. The tumor cells were exclusively BCL-2 positive in 23 (56%) of the 42 specimens tested, consisting of six follicular lymphomas and 17 diffuse lymphomas. A small to moderate number of small lymphocytes, which were positive for CD3 and CD45RO, were intermingled with the neoplastic B cells. In 21 (46%) of the 46 specimens examined, follicular dendritic cell (FDC) networks were easily detectable with monoclonal antibody 1F8, 1B12, or CAN.42, and six diffuse centroblastic lymphomas of the CD5− CD10−, CD20+ phenotype among them contained a remnant of a FDC network. In 27 (66%) of the 41 cases, a few scattered CAN.42-positive mononuclear cells were also observed among the malignant cells and epithelioid cells. These mononuclear cells were often spindle-shaped and negative for CD68. Large tumor cells that exhibited membrane staining were also identified by CAN.42 immunostaining in one (2%) of the 43 cases examined (Case 44).
In Situ Hybridization Detection of EBV Genomes
A positive signal for the EBERs was identified in only a small number of medium- or large-sized nuclei in four of the 25 specimens examined (Cases 4, 17, 29, and 30).
Molecular Genetic Studies
The bcl-2/IgH translocation at the major breakpoint region was detected in six (19%) of the 31 specimens examined (Cases 3, 4, 14, 23, 25, and 46).
Clinical Findings
None of the patients exhibited any manifestations of systemic sarcoidosis or had undergone lymphangiography before the lymph node biopsy. The patients consisted of 18 males and 32 females. They ranged in age from 31 to 88 years old, with a mean age of 66 years and a median age of 71 years. All 50 patients initially presented with lymphadenopathy, although there was evidence of extranodal disease in 19. The extranodal sites consisted of bone marrow (n = 6), epipharynx (n = 4), spleen (n = 3), spleen and pleura (n = 1), liver and spleen (n = 1), skin (n = 1), breast and bone marrow(n = 1), tonsil (n = 1), and stomach and lung (n = 1). Bulky masses were observed in 27 cases (54%) and were most common in the neck (n = 17), followed by inguinal (n = 5), axillary (n = 4), intra-abdominal (n = 1), and chest wall (n = 1) lymph nodes. “B” symptoms, such as fever or body weight loss, were recorded in only seven cases (14%).
Twenty-eight cases (56%) had localized lesions (Stage I or II), and 22 (44%) had advanced disease (Stage III or IV). At the time of diagnosis, five patients had chronic thyroiditis (Cases 6, 14, 22, 24, and 37), three had rheumatoid arthritis (Cases 33, 38, and 40), one had antiphospholipid antibody syndrome (Case 7), one had diabetic nephropathy (Case 11), and one had prostatic adenocarcinoma (Case 49). Rheumatoid factor was positive in two cases (Cases 10 and 18), and antinuclear antibody was positive in one (Case 39). None of the 50 patients had paraproteinemia or Bence-Jones protein.
Table 2 summarizes the clinical features at presentation (25). In comparison with patients with diffuse large B-cell lymphomas (DLBCL), patients with LBCL-ER showed a higher age distribution (P = .03), a female predominance (P = .001), and a less frequent involvement of extranodal sites >1 (P = .004), but no significant difference in other aggressive clinical parameters such as performace status (PS), serum LDH level, stage, B symptom, or International Prognostic Index (IPI). A comparison with FL also showed that the patients with LBCL-ER were characterized by a higher age distribution (P = .0006), and higher serum LDH level (P = .01), but no significant difference in extranodal involvement. The IPI score was significantly higher in LBCL-ER than that in FL.
Therapeutic Response and Prognosis
Thirty-five patients were treated with combination chemotherapy regimens, for the most part consisting of CHOP (cyclophosphamide, doxarubicin, vincristine, and prednisolone); seven with radiation and chemotherapy, and four with radiotherapy alone; The other four patients received no medication.
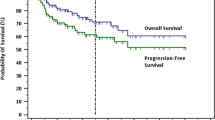
Follow-up information was available in 44 cases, with a follow-up time of one to 182 months (mean, 35 mo; median 20 mo). One patient (Case 11) who received no medication died of renal failure and was excluded from the survival analyses. After excluding this one case, the overall survival rate of the other 43 cases was 60% at 5 years, and 40% at 8 years. LBCL-ER thus showed a survival curve almost identical with that of DLBCL (Fig. 2) and significantly inferior to that for FL (Fig. 3). No significant difference in survival was found by the growth pattern (nodular and diffuse), CD10 positivity, BCL-2 expression, or the presence or absence of the bcl-2/IgH translocation.

Overall survival for patients with CB and CBCCEP+ and DLBCLEP−. The survival curves of both CB and CBCCEP+ and DLBCLEP− are identical.

Overall survival for patients with CB and CBCCEP+ and FLCLEP−. The CBCCEP+ had a significantly poorer overall survival than those of FLCLEP−.
Univariate Cox analysis identified the following prognostic factors for the entire 217 patients with LBCL-ER and DLBCL: age, PS, serum LDH level, clinical stage, extranodal involvement, presence of B symptoma, and IPI category, but not epithelioid cell response (Table 3). On the other hand, the analysis for 144 patients with LBCL-ER and FL showed an epithelioid cell response, but not extranodal involvement, to be a significant and prognostic factor.
DISCUSSION
Many of the 50 cases in this study shared several clinicopathologic characteristics: that is, (1) 33 (66%) of the patients were >60 years old (median age: 71 y), and there was a female predominance (male to female ratio: 0.6); (2) the tumor was generally of nodal origin with infrequent involvement of the spleen, bone marrow, and pharynx; (3) 27 (54%) of the patients presented with a bulky mass of affected lymph nodes; and (4) the epithelioid cells were accumulated in large demarcated masses, imparting a lymphoepithelioid-(Lennert) lymphoma (LeL)-like appearance to the lymph node lesions. A minority (18%) of the cases in this series were associated with autoimmune diseases. The occurrence of this combination of features in this series was not sufficiently emphasized in our previous report (22), although this variant has been mentioned as an unusual tumor with epithelioid response.
We previously suggested that the majority of this variant of B-cell lymphoma may be of germinal center cell origin (22). In the present study, the CD5− CD10+ phenotype was identified in 43% of the cases, and remnants of follicular dendritic cell (FDC) networks were detected in 46% (26). BCL-2 expression was found in 56% of the cases, a level similar to the 50% BCL-2 positivity in previous study (27). The BCL-2 gene rearrangement was detected in only 19% of the cases examined. Some studies have described a relatively low incidence of BCL-2 gene rearrangement in both conventional diffuse centroblastic lymphoma and follicular lymphoma in Asian countries compared with Western countries, although it was extremely difficult to draw any definitive conclusion from the limited number of cases (28, 29, 30, 31, 32).
This study presented here demonstrated a significant difference in age distribution, sex ratio, and extranodal involvement of more than one site between LBCL-ER and DLBCL. In comparison with FL, LBCL-ER was more closely associated with many aggressive clinical features or parameters (age, PS, serum LDH level, and IPI), but not extranodal involvement or clinical stage. The survival curve of LBCL-ER was identical with that of DLBCL, and significantly inferior to that of FL. These combined clinicopathologic, immunohistologic, and genotypic features of LBCL-ER appeared to be relatively homogeneous as one group, were generally in keeping with those of DLBCL, and simultaneously suggested the possibility that this lymphoma may be derived from the morphological progression of FL.
The issue of lymphoma progression was intensively reviewed by Muller-Hermelink et al. (33). They indicated that histological variability and clinical relevance in morphological progression of FL are still controversial and that patterns of lymphoma progression previously described are not necessarily reproducible today. The marked epithelioid cell response in large B cell lymphoma documented here is easily recognizable, and may be regarded as the landmark for the diagnosis of this lymphoma overlapping morphological FL progression. Further investigations are needed to clarify this issue. Promising results have recently been provided by microarray analysis of the expression of thousands of genes on microchips (34, 35). Alizadeh et al. reported two major groups of diffuse large B-cell lymphomas (DLBCLs) based on gene expression patterns indicative of different stages of B-cell differentiation (36). One major group expresses genes similar to those of germinal center B-cells, and the other expresses genes similar to those of activated B cells. The patients in the former group had significantly better overall survival than did the patients in the latter group (five-year survival of 76% versus 16%; 36). DNA microarray studies are required to classify the genetic features of the LBCL-ER described here.
Recent clinical studies have revealed that peripheral T cell lymphomas other than anaplastic large cell lymphomas have a less favorable outcome than centroblastic lymphomas (37, 38). The five-year overall survival rate for the former is only 25%, and significantly poorer than that (approximately 50%) of the latter (37, 38, 39). This implies that it is of practical importance to make a distinction between lymphoepithelioid lymphoma (LeL) and LBCL-ER (2, 39, 40). In LeL, small lymphocytes with round or somewhat irregular nuclei are the predominant cell form distributed over the entire lesion, although some medium-sized lymphoid cells and immunoblasts are always present. These tumor cells express the helper T cell phenotype. Epithelioid cells are usually accumulated in small, well-defined clusters. Conspicuous fibrosis of the tumor is absent. These features can be regarded as the landmarks that distinguish LeLs from the present cases.
Only a few cases of T cell/histiocyte-rich B-cell lymphomas (TCRBCL) have been reported to exhibit a LeL-like appearance in the literature (10, 13, 18). Among the 50 cases in the present study, reactive T cells constituted only a minor component of the total lymphoid cell population. In addition, the clinical findings of TCRBCLs might be different from those of the present 50 cases. The affected lymph nodes were described as a bulky mass in only 7 (4%) of the 175 cases of TCRBCL (5, 6, 7, 8, 9, 11, 12, 13, 14, 16, 18, 19). The advanced stage of disease may be more common in TCRBCL than in the present series (8, 9, 11, 12, 13, 14, 18, 19) and an association with autoimmune disease also has been recorded in only two (1%) of 175 TCRBCL patients (5, 6, 7, 8, 9, 11, 12, 13, 14, 16, 18, 19).
Patsouris et al. (21) reported 36 cases of immunocytoma with a high epithelioid cell content that had arisen from lymph nodes. The present cases were distinct from those in that they showed no plasmacytoid differentiation, although intracytoplasmic immunoglobulin was demonstrated in 16% of the cases examined. About half (46%) of the series described by Patsouris et al. (21) was associated with autoimmune diseases, mainly Sjögren’s syndrome, whereas nine (18%) of the patients in the present series also had an autoimmune disease. Both tumors mainly occur in elderly females and infrequently involve extranodal sites. The interrelationship and/or clinical similarities between them should be further investigated in the future.
The epithelioid cell response is frequently observed in association with the proliferation of the germinal center and/or monocytoid B cells in the granulomatous lymphadenitis such as cat-scratch disease, toxoplasmosis, and tuberculosis (41). However, its biologic significance in B-cell proliferation remains to be elucidated.
In the present study, CAN.42 immunostaining highlighted scattered mononuclear cells situated among the tumor cells and epithelioid cells, in addition to the FDC network. These mononuclear cells were unreactive with CD21, CD23, and CD68, as described by Raymond et al. (42). They also stated that the tumor cells were positive for CAN.42 antibody in six (5%) of 129 cases of B cell lymphomas (42). In the present study, tumor cells were stained with CAN.42 antibody in only 1 (2%) of the 41 cases examined.
In conclusion, centroblastic and centroblastic/centrocytic lymphomas associated with prominent epithelioid cell response comprise a morphological variant of diffuse large B-cell lymphoma that share a distinct capability of attracting an intense epithelioid cell infiltration and account for a certain proportion of cases of follicular lymphoma progression.
References
Hui PK, Feller AC, Lennert K . High-grade non-Hodgkin’s lymphoma of B-cell type.I. Histopathology. Histopathology 1988; 12: 127–143.
Lennert K, Feller AC . Histopathology of non-Hodgkin’s lymphoma (based on the updated Kiel classification). 2nd ed. Heidelberg, Germany: Springer-Verlag; 1992.
Jaffe ES, Harris NL, Stein H, Vardiman J, editors. Pathology and genetics of tumors of haematopoietic and lymphoid tissues.In: Jaffe ES, Harris NL, Stein H, Vardiman J, editors. The World Health Organization classification of tumors. Lyon, France: IARC Press; 2001.
Warnke RF, Weiss LM, Chan JKC, Clearre ML, Dorfman RF . Tumor of the lymph nodes and spleen.In: Atlas of tumor pathology.3rd series, Fascicle 14. Bethesda, MD: Armed Forces Institute of Pathology; 1995.
Ramsay AD, Smith WJ, Isaacson PG . T-cell-rich B-cell lymphoma. Am J Surg Pathol 1988; 12: 433–443.
Ng CS, Chan JKC, Hui PK, Lau WH . Large B-cell lymphomas with a high content of reactive T cells. Hum Pathol 1989; 20: 1145–1154.
Osborne BM, Butler JJ, Pugh WC . The value of immunophenotyping on paraffin sections in the identification of T-cell rich B-cell large-cell lymphomas: lineage confirmed by JH rearrangement. Am J Surg Pathol 1990; 14: 933–938.
Chittal SM, Brousset P, Voigt J-J, Delsol G . Large B-cell lymphoma rich in T-cells and simulating Hodgkin’s disease. Histopathology 1991; 19: 211–220.
Dalabie J, Vandenberghe E, Kennes C, Verhoef G, Foschini MP, et al. Histiocyte-rich B-cell lymphoma. A distinct clinicopathologic entity possibly related to lymphocyte predominant Hodgkin’s disease, paragranuloma subtype. Am J Surg Pathol 1992; 16: 37–48.
Macon WR, Williams ME, Greer JP, Stein RS, Collins RD, Cousar JP . T-cell-rich B-cell lymphomas. A clinicopathologic study of 19 cases. Am J Surg Pathol 1992; 16: 351–363.
Rodriguez J, Pugh WC, Cabanillas F . T-cell-rich B-cell lymphoma. Blood; 1993; 82: 1586–1589.
Krishnan J, Wallberg K, Frizzera G . T-cell-rich large B-cell lymphoma. A study of 30 cases, supporting its histologic heterogeneity and lack of clinical distinctiveness. Am J Surg Pathol 1994; 18: 455–465.
Baddoura FK, Chan WC, Masih AS, Mitchell D, San NCJ, Weisenburger DD . T-cell-rich B-cell lymphoma. A clinicopathologic study of eight cases. Am J Clin Pathol 1995; 103: 65–75.
Greer JP, Macon WR, Lamar RE, Wolff SN, Stein RS, Flexner SJM, et al. T-cell rich B-cell lymphomas: diagnosis and response to therapy of 44 patients. J Clin Oncol 1995; 13: 1742–1750.
de Jung D, Van Gorp J, Sie-Go D, Van Heerde P . T-cell rich B-cell non-Hodgkin’s lymphoma. a progressed form of follicle center cell lymphoma and lymphocyte predominance Hodgkin’s disease. Histopathology 1996; 28: 15–24.
McBride JA, Rodriguez J, Luthra R, Ordóñez NG, Cabanillas F, Pugh WC . T-cell-rich B large-cell lymphoma simulating lymphocyte-rich Hodgkin’s disease. Am J Surg Pathol 1996; 20: 193–201.
Schmidt U, Herbst J, Metz KA, Leder L-D . How to differentiate between T-cell-rich B-cell lymphoma and lymphocyte-predominant Hodgkin’s disease: evidence for the value of MB1 and 4KB5 immunostaining. J Pathol 1996; 179: 138–144.
Camilleri-Bröet S, Molina T, Audouin J, Le Tourneau A, Diebold J . Morphological variability of tumour cells in T-cell-rich B-cell lymphoma. A histopathological study of 14 cases. Virchows Arch 1996; 429: 243–248.
Skinnider BF, Connors JM, Gastcoyne RD . Bone marrow involvement in T-cell-rich-B-cell lymphoma. Am J Clin Pathol 1997; 108: 570–578.
Rüdiger T, Ott G, Ott MM, Müller-Deubert SM, Müller-Hermelink HK . Differential diagnosis between classic Hodgkin’s lymphoma, T-cell-rich B-cell lymphoma, and paragranuloma by paraffin immunohistochemistry. Am J Surg Pathol 1999; 22: 1184–1191.
Patsouris E, Noël H, Lennert K . Lymphoplasmacytic/lymphoplasmacytoid immunocytoma with a high content of epithelioid cells. Histologic and immunohistochemical findings. Am J Surg Pathol 1990; 14: 660–670.
Kojima M, Nakamura S, Motoori T, Kurabayashi Y, Hosomura Y, Itoh H, et al. Centroblastic and centroblastic-centrocytic lymphomas associated with prominent epithelioid granulomatous response without plasma cell differentiation: a clinicopathologic study of twelve cases. Hum Pathol 1996; 27: 660–667.
Carbone PP, Kaplan HS, Musshoff K, Smithers DW, Tubiana M . Report of the committee on Hodgkin’s disease staging classification. Cancer Res 1971; 31: 1860–1861.
Elenitoba-Johnson KS, Kumar S, Lim MS, Kignima DW, Raffeld M, Jaffe ES . Marginal zone B-cell lymphoma with monocytoid B-cell lymphocytes in pediatric patients without immunodeficiency: a report of two cases. Am J Clin Pathol 1997; 107: 92–98.
The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. A predictive model for aggressive non-Hodgkin’s lymphoma. N Engl J Med 1993; 329: 987–994.
Scoazec JY, Berger F, Mogaud JP, Brochier J, Coiffier B, Bryon P-A . The dendritic reticulum cell pattern in B cell lymphomas of the small cleaved, mixed and large cell types. An immunohistochemical study of 48 cases. Hum Pathol 1989; 20: 124–131.
Schnnider BF, Horsman DE, Dupus B, et al. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999; 30: 803–808.
Loke SL, Pittaluga S, Srivastava G, Reffeld M, Ho FCS . Translocation of bcl-2 gene in non-Hodgkin’s lymphomas in Hong Kong Chinese. Br J Haematol 1990; 76: 65–69.
Liang R, Chan V, Chan TK, Chiyu E, Todd D . Rearrangement of immunoglobulin T-cell receptor, and bcl-2 genes in malignant lymphomas in Hong Kong. Cancer 1990; 66: 1743–1747.
Takechi M, Tanaka K, Hashiomoto T, Asaoku H, Dohy H, et al. Cytogenetic, molecular biological and clinical study of B-cell lymphomas with 14;18 translocation in Japanese patients. Leukemia 1991; 5: 1069–1075.
Fang J-M, Finn WG, Hussong JW, Goolsby CL, Cubbon AS, Variakojis D . CD10 antigen expression correlates with the t(14;18)(q32;q21) major breakpoint region in diffuse large B-cell lymphoma. Mod Pathol 1999; 12: 295–300.
Akasaka T, Akasaka H, Ueda C, Yonetani N, Masesako Y, et al. Molecular and clinical features of non-Burkitt’s diffuse large-cell lymphoma of B-cell type associated with the c-MYC/immunoglobulin heavy-chain fusion gene. J Clin Oncol 2000; 18: 510–518.
Muller-Hermelink HK, Zettl A, Pfeifer W, Ott G . Pathology of lymphoma progression. Histopathology 2001; 38: 285–306.
Friend SH . How DNA microassays and expression profiling will affect clinical practice. BMJ 1999; 319: 1306–1307.
Stewart DJ . Masking and using microassays: a short course at Cold Spring Harbor Laboratory. Genome Res 2000; 10: 1–3.
Allzadeh AA, Elsen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct type of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000; 40: 3503–3511.
The Non-Hodgkin’s Lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood 1997; 89: 3909–3918.
Melnyk A, Rodriguez A, Pugh WC, Cabannillas F . Evaluation of the revised European-American Lymphoma Classification confirms the clinical relevance of immunophenotype in 560 cases of aggressive non-Hodgkin’s lymphoma. Blood 1997; 89: 4514–4520.
Nakamura S, Suchi T . A clinicopathologic study of node-based, low-grade, peripheral T-cell lymphoma. Angioimmunoblastic lymphoma, T-zone lymphoma, and lymphoepithelioid lymphoma. Cancer 1992; 69: 2565–2578.
Patsouris E, Noël H, Lennert K . Histological and immunohistological findings in lymphoepithelioid cell lymphoma (Lennert’s lymphoma). Am J Surg Pathol 1988; 12: 341–350.
Kojima M, Nakamura S, Itoh H, Yoshida K, Shimizu K, Motoori T, et al. Occurrence of monocytoid B-cells in reactive lymph node lesions. Pathol Res Pract 1998; 194: 559–565.
Raymond I, Saati TA, Tkaczuk J, Chittal S, Delsol G . CAN.42, a new monoclonal antibody directed against a fixative-resistant antigen of follicular dendritic reticulum cells. Am J Pathol 1997; 151: 1577–1585.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kojima, M., Nakamura, S., Ichimura, K. et al. Centroblastic and Centroblastic/Centrocytic Lymphoma Associated with a Prominent Epithelioid Granulomatous Response: a Clinicopathologic Study of 50 Cases. Mod Pathol 15, 750–758 (2002). https://doi.org/10.1097/01.MP.0000018980.83088.D3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000018980.83088.D3
