
Abstract
Tuberous sclerosis complex (TSC) is an autosomal-dominant disorder characterized by seizures, mental retardation, autism, and tumors of multiple organs. Renal disease in TSC includes angiomyolipomas, cysts, and renal cell carcinomas. It is known that somatic mutations in the von Hippel Lindau (VHL) tumor suppressor gene occur in most clear cell renal carcinomas. To determine whether TSC-associated clear cell carcinomas also contain VHL mutations, we analyzed six tumors for loss of heterozygosity in the VHL gene region of chromosome 3p and for mutations in the VHL gene. Four of the patients were women between the ages of 34 and 68 years, and two were males under the age of 21 years. The loss of heterozygosity analysis was performed using polymorphic microsatellite markers, and the mutational analysis was performed using direct sequencing. Chromosome 3p loss of heterozygosity was not detected, and no VHL mutations were identified. These findings suggest that mutations in the TSC1 and TSC2 genes lead to clear cell renal carcinogenesis via an alternate pathway not involving VHL mutations.
Similar content being viewed by others
INTRODUCTION
Tuberous sclerosis (TSC) is a tumor suppressor gene syndrome characterized by seizures, mental retardation, autism, and tumors in the brain, retina, kidney, heart, and skin (1). The birth incidence of TSC is estimated to be 1 in 11,000 (2). TSC is transmitted with an autosomal-dominant pattern of inheritance with 95% penetrance. Sixty percent of TSC patients have apparent new mutations with no prior family history of the disease (3). TSC affects nearly every major organ system. The most common causes of death in TSC patients are renal disease, brain tumors, and lung disease (4).
Renal disease in TSC includes cysts, angiomyolipomas, and renal cell carcinoma. Angiomyolipomas are benign tumors with vascular, smooth muscle, and lipomatous components (5). Renal cell carcinoma has been reported in young children, as well as adults, with TSC and has an average age at onset of about 33 years (6, 7, 8, 9). Renal cell carcinoma in TSC is pathologically heterogeneous, including clear cell, papillary, and chromophobe types (7, 8). This heterogeneity contrasts with renal cell carcinoma in von Hippel Lindau (VHL) disease or hereditary papillary renal carcinoma (HPRC), which are associated with clear cell and papillary tumors, respectively. Clear cell renal carcinoma appears to be the most common histologic subtype in TSC patients (10).
Mutations in the VHL gene occur in most sporadic clear cell renal carcinomas (11, 12, 13). In this study, we analyzed six clear cell renal carcinomas for mutations in the VHL gene and for loss of heterozygosity in the VHL gene region of chromosome 3p. We reasoned that if VHL mutations were found, it would indicate that mutations in VHL are involved in clear cell renal carcinogenesis in TSC patients. If mutations were not found, it would strongly suggest the presence of an alternate pathway for clear cell renal carcinogenesis involving the TSC1 and TSC2 genes.
METHODS
Patients and Tumor Specimens
All patients had clinical diagnoses of TSC, as defined by the 1998 Tuberous Sclerosis Complex Consensus Conference (14). Their underlying TSC1 or TSC2 mutations were not known. All underwent nephrectomy because of lesions suspicious for renal cell carcinoma, with the exception of Patient 354. Patient 354 has dialysis-dependent end-stage renal failure and underwent nephrectomy because of painful polycystic kidneys. His renal cell carcinoma was an incidental finding.
Pathologic Analysis
Paraffin sections of each tumor were stained with hematoxylin and eosin for pathologic review. Immunohistochemistry was performed using AE1/AE3 cytokeratin antibodies and HMB-45 antibody (both from Biogenex, San Ramon, CA). Endogenous peroxidase activity was quenched by a 30-minute incubation in 0.3% hydrogen peroxide. Slides were blocked in normal goat serum. After incubation with the specific antibodies and with the secondary biotinylated goat anti-mouse antibody (Biogenex), the signal was visualized by incubation with streptavidin-peroxidase and diaminobenzidine (Biogenex).
Molecular Genetic Analysis and Detection of Allelic Loss
DNA was extracted from unstained, paraffin-embedded tumor tissue in 50 mm of KCl, 10 mm of Tris (pH 8.3), 1.5 mm of MgCl2, 100 μg/mL of bovine serum albumin, 0.45% Tween 20, 0.45% NP-40, and 100 μg/mL of proteinase K. After overnight incubation at 65° C, the proteinase K was heat inactivated at 95° C for 10 minutes. A 2-μL aliquot of DNA was used in a 20-μL polymerase chain reaction (PCR). The three coding exons of the VHL gene were examined by direct sequencing in both the forward and reverse directions, using primers that have been previously reported (15). VHL mutational analysis from paraffin tissue has been previously validated (16).
Loss of heterozygosity analysis was performed in tumors and paired normal kidney from patients 353, 618, 622, and 628 using the chromosome 3p marker D3S1478 (Research Genetics, Huntsville, AL). Loss of heterozygosity analysis on chromosome 3p was previously reported for the other two patients (7, 8). A 2.5-μL aliquot of DNA was used in a 10-μL PCR containing 10% glycerol. The PCR amplification consisted of 95° C for 5 minutes, 40 cycles of 95° C for 30 seconds, 55° C for 30 seconds, 72° C for 45 seconds, and a final extension of 72° C for 10 minutes. PCR was performed with [32P]dGTP in the reaction mix. TaqStart antibody (Clontech, Palo Alto, CA) was used in PCR to enhance the specificity. The PCR products for the loss of heterozygosity analysis were resolved by denaturing 8 m urea polyacrylamide gel electrophoresis (Gibco, Grand Island, NY) and visualized by autoradiography.
RESULTS
The patients ranged in age from 11 to 68 years (Table 1). The two younger patients were male, under the age of 21 years. The four older patients were women ranging in age from 34 to 68 years (Table 1). Two of the tumors were included in previous reports of TSC-associated renal cell carcinomas (Patients 120 and 354; 7, 8).
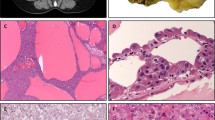
The tumors ranged in maximum dimension from 1.8 to 13 cm. Four of the tumors were Stage I, one was Stage II, and one was Stage III. In four cases, the resected specimen also contained angiomyolipomas, and in five cases, the resected specimen also contained epithelial cysts. Each tumor was centrally reviewed by one of the authors (TA-S) and confirmed to be a clear cell carcinoma and not an epithelioid variant of angiomyolipoma (17); Fig. 1). All of the tumors were positive for cytokeratin and negative for HMB-45. Three tumors were stained for vimentin. The tumors from patients 618 and 628 were vimentin negative, and the tumor from patient 622 was vimentin positive.

Hematoxylin and eosin–stained sections from each tumor. Original magnification, 200×.
Four of the tumors were analyzed for loss of heterozygosity on chromosome 3p as part of this study (Patients 353, 618, 622, and 628), and two tumors had been previously analyzed and reported (7, 8; Patients 120 and 354). Each of the four patients tested as part of this study was heterozygous at the marker D3S1478. Loss of heterozygosity on chromosome 3p was not found in any of the tumors (Table 1).
The coding region of the VHL gene was examined for mutations by PCR amplification, followed by bidirectional DNA sequencing. This was successful for all samples. We did not detect mutations in the VHL gene in any of the tumors (Table 1).
DISCUSSION
Renal cell carcinoma is an uncommon manifestation of TSC (18). However, the occurrence of these cancers in some young children with TSC and in all three rodent models of TSC2 indicates that these carcinomas are specifically associated with TSC. The literature contains numerous case reports and small case series of TSC-associated renal cell carcinomas (6, 7, 8, 10, 19, 20, 21, 22, 23, 24, 25, 26). Malignant transformation of angiomyolipomas has also been described (9).
Animal models provide additional support for a role of the TSC genes in renal carcinogenesis. The Eker rat, which carries a germline mutation in the rat TSC2 homologue, develops renal cysts and carcinomas with an autosomal-dominant pattern of inheritance (27, 28). Two groups have developed mice with targeted inactivation of TSC2 (29, 30). Both mouse models develop renal carcinomas and cysts similar to those of the Eker rat.
In addition to TSC, there are at least two other hereditary forms of renal cell carcinoma in humans for which genes have been identified (31): VHL disease and HPRC. VHL is caused by mutations in the VHL gene (32). HPRC is caused by mutations in the MET gene (33). VHL patients develop almost exclusively renal carcinomas with clear cell morphology, whereas HPRC patients develop almost exclusively papillary tumors. VHL mutations occur in most sporadic clear cell carcinomas (11, 13), but not in other types of renal carcinoma [reviewed in (34)].
In sharp contrast to VHL and HPRC, the renal cell carcinomas in TSC are morphologically heterogeneous, including clear cell, papillary, and chromophobic tumors (7, 8). In this study, we sought to determine whether clear cell renal carcinomas in TSC patients occur independently of mutations in the VHL gene. We analyzed paraffin-embedded tissue specimens from six TSC-associated clear cell renal carcinomas for mutations in the VHL gene and for loss of heterozygosity in the VHL gene region of chromosome 3p. In none of the tumors was loss of heterozygosity in the VHL region or mutations in the VHL gene detected. This is, to our knowledge, the first mutational analysis of any gene in TSC-associated renal carcinomas.
We initially considered two possible models for the development of renal carcinomas in TSC. In the first model, mutations in the TSC genes increase the risk of different pathologic types of renal cell carcinoma, and the development of a clear cell carcinoma involves additional mutational inactivation of the VHL gene. In the second model, mutations in the TSC genes lead to clear cell carcinoma independently of VHL mutations. Our data support the second model and strongly suggest that cellular pathways involving the TSC genes provide an alternate pathway to clear cell renal carcinogenesis. Other studies in humans (35, 36, 37, 38, 39), rodents (40), and dogs (41) have also suggested the existence of pathways leading to clear cell renal carcinogenesis that do not involve mutations in the VHL genes. In contrast to these previous reports, in which the genetic components of the alternate pathways are unknown, all of the patients in our study are known to have TSC.
The fact that TSC patients develop clear cell renal carcinomas indicates that cellular pathways involving the TSC1 and TSC2 genes overlap functionally with pathways involving the VHL gene. Pathways in which tuberin, the TSC2 gene product, is believed to participate include vesicular trafficking (42) and cell cycle regulation (43, 44). Hamartin, the TSC1 gene product, also regulates the cell cycle (45, 46, 47, 48) and affects focal adhesion formation via activation of the GTPase Rho (49). To date, no direct connections between the functions of hamartin and tuberin and the function of the VHL protein, pVHL, have been identified. pVHL targets the hypoxia-inducible transcription factor HIF-1 for degradation, and tumor cells lacking pVHL overproduce HIF target genes, including vascular endothelial growth factor [reviewed in (50)]. pVHL is also believed to play a role in cell cycle regulation and cell differentiation (50).
It is not yet known whether the TSC1 or TSC2 gene is mutated in sporadic renal cell carcinoma. The TSC1 gene is on chromosome 9q34, and chromosome 9q loss of heterozygosity occurs in approximately 30% of sporadic renal cell carcinomas (51). Renal cell carcinomas are known to occur in patients with angiomyolipomas who do not have TSC. In a recent study of 36 renal cell carcinomas associated with angiomyolipomas, 25 of which were from patients without TSC, clear cell was the most common type (10). These sporadic angiomyolipoma-associated renal carcinomas are of particular interest for future mutational analyses of the TSC1 and TSC2 genes.
CONCLUSION
Each year, in the United States alone, renal cell carcinoma is diagnosed in 30,000 people, 10,000 of whom will die of metastatic disease. The majority of these patients have clear cell carcinomas. Renal cell carcinoma is among the most chemotherapy and radiotherapy resistant of all human cancers [reviewed in (52)]. Our data indicate that mutations in the TSC1 or TSC2 genes lead to clear cell renal carcinoma via an alternate pathway that does not involve VHL gene mutations. Elucidating the cellular mechanisms of this alternate pathway may facilitate the identification of novel therapeutic targets for patients with renal cell carcinoma.
References
Gomez M, Sampson JR, Whittemore VH . Tuberous sclerosis complex. New York: Oxford University Press; 1999.
O’Callaghan FJK, Shiell AW, Osborne JP, Martyn CN . Prevalence of tuberous sclerosis estimated by capture-recapture analysis. Lancet 1998; 351: 1490.
Jones AC, Shyamsundar MM, Thomas MW, Maynard J, Idziaszczyk S, Tomkins S, et al. Comprehensive mutation analysis of TSC1 and TSC2 and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet 1999; 64: 1305–1315.
Shepherd CW, Gomez MR, Lie JT, Crowson CS . Causes of death in patients with tuberous sclerosis. Mayo Clin Proc 1991; 66: 792–796.
Eble JN . Angiomyolipoma of kidney. Semin Diagn Pathol 1998; 15: 21–40.
Washecka R, Hanna M . Malignant renal tumors in tuberous sclerosis. Urology 1991; 37: 340–343.
Bjornsson J, Short MP, Kwiatkowski DJ, Henske EP . Tuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic features. Am J Pathol 1996; 149: 1201–1208.
Al-Saleem T, Wessner LL, Scheithauer BW, Patterson K, Roach ES, Dreyer SJ, et al. Malignant tumors of the kidney, brain, and soft tissues in children and young adults with the tuberous sclerosis complex. Cancer 1998; 83: 2208–2216.
Pea M, Bonetti F, Martignoni G, Henske EP, Manfrin E, Colato C, et al. Apparent renal cell carcinomas in tuberous sclerosis are heterogeneous: the identification of malignant epithelioid angiomyolipoma. Am J Surg Pathol 1998; 22: 180–187.
Jimenez RE, Eble JN, Reuter VE, Epstein JI, Folpe AL, de Peralta-Venturina M, et al. Concurrent angiomyolipoma and renal cell neoplasia: a study of 36 cases. Mod Pathol 2001; 14: 157–163.
Shuin T, Kondo K, Torigoe S, Kishida T, Kubota Y, Hosaka M, et al. Frequent somatic mutations and loss of heterozygosity of the von Hippel-Lindau tumor suppressor gene in primary human renal cell carcinomas. Cancer Res 1994; 54: 2852–2855.
Gnarra J, Tory K, Weng Y, Schmidt L, Wei M, Li H, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994; 7: 85–90.
Foster K, Prowse AH, van den Berg A, Fleming S, Hulsbeek MMF, Crossey PA, et al. Somatic mutations of the von Hippel-Lindau disease tumor suppressor gene in non-familial clear cell renal carcinoma. Hum Mol Genet 1994; 3: 2169–2173.
Roach ES, Gomez MR, Northrup H . Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998; 13: 624–628.
Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, et al. Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. J Med Genet 1996; 33: 328–332.
Zhuang Z, Gnarra JR, Dudley CF, Zbar B, Linehan WM, Lubensky IA . Detection of von Hippel-Lindau disease gene mutations in paraffin-embedded sporadic renal cell carcinoma specimens. Mod Pathol 1996; 9: 838–842.
Eble JN, Amin MB, Young RH . Epithelioid angiomyolipoma of the kidney: a report of five cases with a prominent and diagnostically confusing epithelioid smooth muscle component. Am J Surg Pathol 1997; 21: 1123–1130.
Tello R, Blickman JG, Buonomo C, Herrin J . Meta analysis of the relationship between tuberous sclerosis complex and renal cell carcinoma. Eur J Radiol 1998; 27: 131–138.
Weinblatt ME, Kahn E, Kochen J . Renal cell carcinoma in patients with tuberous sclerosis. Pediatrics 1987; 80: 898–903.
Taylor RS, Joseph DB, Kohaut EC, Wilson ER, Bueschen AJ . Renal angiomyolipoma associated with lymph node involvement and renal cell carcinoma in patients with tuberous sclerosis. J Urol 1989; 141: 930–932.
Graves N, Barnes WF . Renal cell carcinoma and angiomyolipoma in tuberous sclerosis: case report. J Urol 1986; 135: 122–123.
Kulkarni S, Uddar M, Deshpande SG, Vaid S, Wadia RS . Renal cell carcinoma as significant manifestation of tuberous sclerosis complex. J Assoc Physicians India 2000; 48: 351–353.
Hidai H, Chiba T, Takagi Y, Taki A, Nagashima Y, Kuroko K . Bilateral chromophobe cell renal carcinoma in tuberous sclerosis complex. Int J Urol 1997; 4: 86–89.
Robertson FM, Cendron M, Klauber GT, Harris BH . Renal cell carcinoma in association with tuberous sclerosis in children. J Pediatr Surg 1996; 31: 729–730.
Aoyama T, Fujikawa K, Yoshimura K, Sasaki M, Itoh T . Bilateral renal cell carcinoma in a patient with tuberous sclerosis. Int J Urol 1996; 3: 150–151.
Sampson JR, Patel A, Mee AD . Multifocal renal cell carcinoma in sibs from a chromosome 9 linked (TSC1) tuberous sclerosis family. J Med Genet 1995; 32: 848–850.
Yeung RS, Xiao GH, Jin F, Lee WC, Testa JR, Knudson AG . Predisposition to renal carcinoma in the Eker rat is determined by germ-line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc Natl Acad Sci U S A 1994; 91: 11413–11416.
Kobayashi R, Hirayama Y, Kobayashi E, Kubo Y, Hino O . A germline insertion in the tuberous sclerosis (Tsc2) gene gives rise to the Eker rat model of dominantly inherited cancer. Nat Genet 1995; 9: 70–74.
Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T . Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res 1999; 59: 1206–1211.
Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ . Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest 1999; 104: 687–695.
Zbar B, Lerman M . Inherited carcinomas of the kidney. Adv Cancer Res 1998; 75: 163–201.
Latif F, Tory K, Gnarra JR, Yao M, Duh F-M, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993; 260: 1317–1320.
Schmidt L, Duh F-H, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 1997; 16: 68–73.
Gnarra JR . von Hippel-Lindau gene mutations in human and rodent renal tumors—association with clear cell phenotype. J Natl Cancer Inst 1998; 90: 1685–1687.
Clifford SC, Prowse AH, Affara NA, Buys CH, Maher ER . Inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: evidence for a VHL-independent pathway in clear cell renal tumourigenesis. Genes Chromosom Cancer 1998; 22: 200–209.
Bos SD, van den Berg E, Dijkhuizen T, van den Berg A, Draaijers TG, Mensink HJ . Genetic analysis of 2 cases of clear cell renal cancer in 2 sisters. Int J Cancer 1998; 77: 494–497.
Teh BT, Giraud S, Sari NF, Hii SI, Bergerat JP, Larsson C, et al. Familial non-VHL non-papillary clear-cell renal cancer. Lancet 1997; 349: 848–849.
Woodward ER, Clifford SC, Astuti D, Affara NA, Maher ER . Familial clear cell renal cell carcinoma (FCRC): clinical features and mutation analysis of the VHL, MET, and CUL2 candidate genes. J Med Genet 2000; 37: 348–353.
Martinez A, Fullwood P, Kondo K, Kishida T, Yao M, Maher ER, et al. Role of chromosome 3p12-p21 tumour suppressor genes in clear cell renal cell carcinoma: analysis of VHL dependent and VHL independent pathways of tumorigenesis. Mol Pathol 2000; 53: 137–144.
Okimoto K, Kouchi M, Kikawa E, Toyosawa K, Koujitani T, Tanaka K, et al. A novel “Nihon” rat model of a Mendelian dominantly inherited renal cell carcinoma. Jpn J Cancer Res 2000; 91: 1096–1099.
Jonasdottir TJ, Mellersh CS, Moe L, Heggebo R, Gamlem H, Ostrander EA, et al. Genetic mapping of a naturally occurring hereditary renal cancer syndrome in dogs. Proc Natl Acad Sci U S A 2000; 97: 4132–4137.
Xiao GH, Shoarinejad F, Jin F, Golemis EA, Yeung RS . The tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein (GAP) in modulating endocytosis. J Biol Chem 1997; 272: 6097–6100.
Soucek T, Pusch O, Wienecke R, DeClue JE, Hengstschlager M . Role of the tuberous sclerosis gene-2 product in cell cycle control. Loss of the tuberous sclerosis gene-2 induces quiescent cells to enter S phase. J Biol Chem 1997; 272: 29301–29308.
Ito N, Rubin GM . gigas, a Drosophila homolog of tuberous sclerosis gene product-2, regulates the cell cycle. Cell 1999; 96: 529–539.
Miloloza A, Rosner M, Nellist M, Halley D, Bernaschek G, Hengstschlager M . The TSC1 gene product, hamartin, negatively regulates cell proliferation. Hum Mol Genet 2000; 9: 1721–1727.
Benvenuto G, Li S, Brown SJ, Braverman R, Vass WC, Cheadle JP, et al. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene 2000; 19: 6306–6316.
Potter CJ, Huang H, Xu T . Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell 2001; 105: 357–368.
Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK . The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell 2001; 105: 345–355.
Lamb RF, Roy C, Diefenbach TJ, Vinters HV, Johnson MW, Jay DG, et al. The TSC1 tumour suppressor hamartin regulates cell adhesion through ERM proteins and the GTPase Rho. Nat Cell Biol 2000; 2: 281–287.
Kondo K, Kaelin WG Jr . The von Hippel-Lindau tumor suppressor gene. Exp Cell Res 2001; 264: 117–125.
Cairns P, Tokino K, Eby Y, Sidransky D . Localization of tumor suppressor loci on chromosome 9 in primary human renal cell carcinomas. Cancer Res 1995; 55: 224–227.
Amato RJ . Chemotherapy for renal cell carcinoma. Semin Oncol 2000; 27: 177–186.
Acknowledgements
This work was supported by grants from the Tuberous Sclerosis Alliance (Silver Spring, MD) and the NIH (RO1 DK51052).
We are grateful to Dr. Paul Cairns for critical review of the manuscript and to Catherine Renner and the Fox Chase Cancer Center Experimental Histopathology Facility for expert assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Duffy, K., Al-Saleem, T., Karbowniczek, M. et al. Mutational Analysis of the von Hippel Lindau Gene in Clear Cell Renal Carcinomas from Tuberous Sclerosis Complex Patients. Mod Pathol 15, 205–210 (2002). https://doi.org/10.1038/modpathol.3880517
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880517
Keywords
This article is cited by
-
Tuberous sclerosis and the kidney: from mesenchyme to epithelium, and beyond
Pediatric Nephrology (2005)
