
Abstract
In a previous study, we demonstrated that the G1 cell cycle checkpoint in carcinomas of the breast is frequently abrogated by loss of p16, the product of the CDKN2/INK4A gene, and, to a lesser extent, by loss of pRB, the product of the retinoblastoma gene. The purpose of the present study was to determine whether other mechanisms of cell cycle deregulation exist in breast cancers which have retained RB and p16 function. Paraffin sections of 81 invasive breast carcinomas (49 ductal, 26 lobular, 6 mixed) were reacted with monoclonal antibodies against cyclin D1 and p53, using optimized immunohistochemical staining protocols. The staining results were correlated with the expression of p16 and pRB, and with a variety of pathological parameters and DNA ploidy. Twenty-five tumors (31%) accumulated (presumably mutant) p53 and 28 (35%) overexpressed cyclin D1; 7 carcinomas (not including any pure lobular cancers) abnormally expressed both proteins. p53 accumulation correlated with nuclear, mitotic, and overall grade, but not with tumor size, lymph node involvement, or DNA ploidy. Overexpression of cyclin D1 was not associated with any of the patho-biological variables. There was an inverse correlation between loss of p16 and high levels of p53, but not cyclin D1. The G1 cell cycle checkpoint, which is controlled by RB, cyclin D1, and p16, was abrogated in 65% of carcinomas, and only p53 was abnormal in an additional 17%. The number of abnormally expressed genes correlated with mitotic activity and overall tumor grade, but not with tumor histology, size, or nodal status, suggesting that cell cycle deregulation is an early event in breast tumorigenesis. Only 18% of the carcinomas showed a normal level of expression of the four genes tested, and p16 appeared to be the most common target of cell cycle deregulation. These data point to the importance of cell cycle regulatory protein abnormalities in human breast cancer.
Similar content being viewed by others

INTRODUCTION
It is generally accepted that several pathways need to be abrogated in the genesis of a malignant neoplasm. There is an increasing body of evidence that deregulation of one or more of the cell cycle checkpoints is among the most common abnormalities in human neoplasia. We have been particularly interested in the genes controlling the late G1 restriction point. Possibly the most critical component of the latter is pRB, the product of the retinoblastoma gene. Hyperphosphorylation, and hence inactivation of pRB is catalyzed by cyclin dependent kinases (CDKs), most notably CDK4 and 6. The CDKs are activated by binding to their respective cyclins, including cyclin D1 (1). They are inhibited by CDK inhibitors including p15INK4B and p16INK4A (specific for CDK4/6) and p21WAF1. The latter, in turn, is partially under the transcriptional control of p53 (2).
We recently demonstrated that abnormal expression of the genes controlling the G1 restriction point is a very common occurrence in carcinomas of the lung and bladder (3, 4). These abnormalities are associated with a variety of clinico-pathologic features and may be of prognostic significance. In an earlier study, we showed that some 50% of breast cancers were negative for p16, and a smaller number were pRB negative (5). We are particularly interested in the molecular differences between so-called ductal and lobular carcinomas; thus, the latter were purposely over-represented in this series. In the present follow-up study, we wanted to test the hypothesis that in those tumors that had retained both pRB and p16, the G1 cell cycle checkpoint might be abrogated by abnormal expression of two other important genes, specifically cyclin D1 and p53. Cyclin D1 has been reported to be overexpressed in some 25 to 75% of breast cancers, and this may be associated with better survival (6). Accumulation of p53, which occurs in 10 to 60% of breast cancers, is usually taken as a surrogate marker of a mutation in the gene. However, it is well recognized that the concordance between level of the protein and abnormalities at the DNA level is by no means perfect (7, 8, 9). We wished to correlate the expression patterns of cyclin D1 and p53 in breast cancer with important pathological features and with DNA ploidy, which is a weak but significant prognostic indicator, particularly in node-negative disease (10). In conjunction with our previously reported pRB and p16 expression data, we were able to comprehensively evaluate the expression pattern of many of the genes controlling the G1 restriction point in the same series of breast cancers. The results show that the cell cycle checkpoint was indeed abrogated in the great majority of cases, and these abnormalities correlated with the proliferative activity of the tumors but not with pathological indicators of metastatic potential. In addition, they were relatively evenly distributed between ductal and lobular carcinomas.
MATERIALS AND METHODS
Tissues
We previously reported pRB and p16 staining results on 104 primary breast cancers (5). Of those, 79 cases had sufficient material for further studies. We added two cases that had not been studied before, for a total of 81. All tumors were from the surgical pathology files of the University of North Carolina Hospitals and had been accessioned between 1987 and 1995. The selection criteria were previously described (5). One or two representative paraffin blocks were utilized from each tumor. Because p53 antigenicity may be spuriously lost in paraffin sections stored at room temperature (11), we used test and control slides that had been stored at 4° C. Because we are particularly interested in the biological differences between ductal and lobular carcinomas, the latter were purposely over-represented. Most of the lobular cancers were of the classical type. A small number of tumors showing both lobular and ductal differentiation were also included in this study. The grading of all carcinomas followed the Nottingham modification of the Scarff-Bloom-Richardson scoring system (12). Review of the original pathology reports yielded tumor size, axillary lymph node status, and DNA ploidy in many, but not all cases. The S-phase fraction could not be reliably determined in a sufficient number of cases. Because biochemical estrogen and progesterone receptor data were available for less than half of the tumors, these data were not included in our analysis.
Reagents
Rabbit polyclonal and murine monoclonal (clone G175–405) anti-p16 antibodies were obtained from PharMingen (San Diego, CA). Murine monoclonal anti-p53 antibody (clone DO7) was purchased from Novocastra/Vector (Burlingame, CA). Murine monoclonal anti-human cyclin D1 antibody (clone DCS-6) was obtained from Lab Vision/NeoMarkers (Freemont, CA). Nonspecific mouse IgG1 was used as a negative control. The Vectastain Elite ABC kit from Vector was used for the detection reactions.
Immunohistochemistry
The immunohistochemical assays for p16 using the polyclonal and monoclonal antibodies, respectively, were recently described in detail (3). Briefly, the polyclonal antibody was used at a 1:400 dilution at 4° C, and the monoclonal antibody at 2 μg/mL at room temperature (RT) overnight. An antigen retrieval step (see below) preceded the p16 monoclonal antibody incubation.
Standard ABC peroxidase techniques were used to demonstrate the presence of cyclin D1 or p53 in formalin-fixed, paraffin-embedded tissues. Four-μm paraffin sections were incubated for 30 min in 0.01 m citrate buffer (95 to 100° C), and then incubated with anti-p53 antibody DO7 diluted 1:500 for 25 min at RT. For detection of cyclin D1 expression, sections were incubated with antibody DCS-6 diluted 1:30 for 2 hours at RT after antigen retrieval in hot citrate buffer. Negative control slides were treated with nonspecific mouse IgG1 at equivalent conditions. The detection reactions for p53 and cyclin D1 utilized the Vectastain Elite ABC kit using conditions recommended by the manufacturer. Diaminobenzidine with hematoxylin counterstain was used for color development.
The following external positive controls were used. A small cell carcinoma of the lung known to overexpress p16 and p53 was utilized as a positive control for both proteins. A cyclin D1 overexpressing parathyroid adenoma served as a positive control for cyclin D1. In addition, a cell block of lung cancer cell line NCI-H441 was used as a positive control for both p53 (missense mutation) and cyclin D1 (overexpression on Western blot).
Interpretation of Immunostains
Our simple dichotomous scoring system for pRB and p16 has been described elsewhere (3, 13). Abnormal protein expression was evidenced by loss of nuclear staining specifically in the neoplastic cells, with preserved reactivity in admixed normal cells.
In some cases, nuclear staining for p53 or cyclin D1 could be observed in a small number of benign epithelial and stromal cells. Thus, a cutoff value of 5% nuclear staining was chosen to separate the normal (negative) from the abnormal (positive) tumors. Cytoplasmic reactivity, if present, was disregarded. Some carcinomas had a small number of scattered positive cells, comparable to the distribution in adjacent benign tissue, and these were considered normal.
Statistical Analysis
To probe for statistically significant correlations between variables, χ2 analysis and Fisher’s exact test were performed. A P value of less than .05 was considered statistically significant.
RESULTS
Staining Patterns
In our initial report, 18 of 104 breast cancers were inconclusive with regard to their pRB or p16 expression patterns (5). Alternate paraffin blocks of 11 of these tumors could be reanalyzed. The single pRB inconclusive tumor was found to be positive on reanalysis. Using the p16 polyclonal antibody, seven previously unevaluable breast cancers were found to be positive and three were negative. Because of our recent observation that the p16 polyclonal antibody may give false positive staining results (14), 37 of 45 cases that were positive for both p16 and pRB on initial analysis were reanalyzed with the specific anti-p16 monoclonal antibody. Thirty-one tumors remained p16 positive, three were negative, and three were inconclusive. A consensus p16 expression score could be obtained in 77 of the 81 cases included in this study. The remaining four cases either showed nonspecific loss of antigenicity or had unacceptably high background staining. Almost half of the interpretable cases were p16 negative, in agreement with our earlier results.
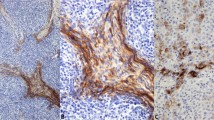
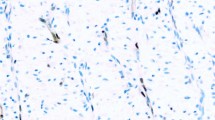
Eighty-one cases were evaluated for p53 and cyclin D1. Twenty-five tumors (31%) had elevated levels of p53 and 28 tumors (35%) overexpressed cyclin D1. In most cases with p53 or cyclin D1 overexpression, there was nuclear staining throughout the tumor (Figs. 1A and 2A). However, rare tumors showed distinctly heterogeneous staining for p53 or cyclin D1 in that the proteins were clearly expressed in some areas of the lesion, but not in others (Fig. 1B). In one ductal carcinoma, expression of cyclin D1 correlated with the biphasic morphology of the tumor: the poorly differentiated areas stained for cyclin D1, whereas the better differentiated areas were negative (Fig. 2, B-C).

Abnormal p53 expression patterns. A, ductal carcinoma with diffuse overexpression of p53. Almost all tumor cells show strong nuclear staining. B, low-grade ductal carcinoma with focal p53 accumulation (intratumor heterogeneity). The neoplastic ducts on the right (arrows) show strong nuclear staining, while the neoplastic ducts on the left (arrowheads) are negative.

Abnormal cyclin D1 expression patterns. A, ductal carcinoma with diffuse overexpression of cyclin D1. The majority of tumor cells show strong nuclear and weaker cytoplasmic staining. B and C, ductal carcinoma with focal cyclin D1 overexpression (intratumor heterogeneity). In the poorly differentiated areas of the lesion, strong staining is observed (B). In the better differentiated areas, the neoplastic cells do not stain (C).
Loss of p16 and Overexpression of p53 and Cyclin D1 Stratified by Patho-Biological Features
Because our review of the surgical pathology records yielded further information, we were able to correlate the loss of p16 expression with additional parameters that were not available at the time of our initial study (5). Abnormal p16 expression was not associated with axillary lymph node involvement or DNA aneuploidy. However, it tended to be more common in tumors larger than 2 cm (P = .08).
We were interested in the question whether abnormal expression of p53, cyclin D1, or both was correlated with a number of histopathological tumor characteristics. There was no statistically significant difference in the expression pattern of the two proteins between invasive ductal and lobular carcinomas, although we did not find coaccumulation of p53 and cyclin D1 in any of the 26 lobular cancers in this study (Table 1). p53 positive tumors were characterized by higher nuclear, mitotic, and overall grades, but were not significantly larger, more frequently metastatic, or DNA aneuploid, compared to p53 negative cases (Table 2). In contrast, cyclin D1 overexpression was not associated with DNA ploidy or any of the pathological parameters studied (Table 2).
Relationship Between Cell Cycle Regulatory Proteins
We and others previously reported the strict inverse relationship between loss of p16 and pRB in breast cancer (5, 15). Because the number of pRB negative tumors in this study was small, no attempt was made to correlate pRB expression with p53 and cyclin D1 expression patterns. However, we found a strong inverse correlation between p16 negativity and p53 accumulation (P = .007), i.e., abnormal expression of both tumor suppressor genes in the same cancer was an uncommon occurrence (Table 3). In contrast, there was no statistically significant relationship between p16 loss and cyclin D1 overexpression, suggesting that these two events may cooperate in cell cycle deregulation in a significant subset of breast cancers (Table 3). Likewise, there was no association between cyclin D1 and p53 overexpression, except that co-overexpression of cyclin D1 and p53 was significantly less common in tumors of low versus those of intermediate or high nuclear grade (0/37 versus 7/37, P = .014).
Correlation of Abnormal Expression of Four Cell Cycle Regulatory Proteins (pRB, p16, cyclin D1, p53) with Patho-Biological Features
In conjunction with our previously published data on pRB and p16 expression in this cohort of breast cancers, our new data on p53 and cyclin D1 expression abnormalities allowed us to comprehensively study the association between cell cycle deregulation and pathologic features. Abnormal cell cycle regulatory protein expression was common in the 77 breast cancers that could be evaluated for all four markers. Half of the tumors were negative for pRB or p16, as previously reported. Considering cyclin D1, two-thirds of the breast cancers showed an abnormal expression pattern of at least one of the three proteins closely associated with the G1 restriction point (Table 4). If p53 accumulation was included, 63 of 77 breast cancers (82%) showed immunohistochemical evidence of cell cycle deregulation (Table 4). Interestingly, there was no statistically significant difference in the frequency of abnormal cell cycle protein expression between ductal and lobular carcinomas (Table 4). Over 50% of breast cancers abnormally expressed one of the four genes, half as many abnormally expressed two, and only three tumors showed evidence of deregulation of three genes (Table 5). As may be expected, the mitotic activity of the tumors was strongly associated with the number of abnormally expressed genes (Table 5). Tumors with intermediate or high mitotic activity had at least one deregulated gene; the 14 “normal” cases all had low mitotic indices. Conversely, the three most abnormal cases had high proliferative activity. Overall tumor grade was also correlated with abnormal gene expression. In contrast, the number of abnormally expressed genes was not associated with architectural or nuclear grade, tumor size, or metastasis to axillary lymph nodes (Table 5).
DISCUSSION
There is a growing consensus that one feature common to all malignant human neoplasms is abrogation of cell cycle control. The cell cycle has several restriction points that are controlled by many regulatory genes including cyclins, CDKs, and CDK inhibitors. The restriction point at the G1/S boundary is comparatively well understood. It is principally controlled by the pRB-CDK4/6-cyclin D1-p15INK4B/p16INK4A pathway. We previously showed that two critical components of this pathway, pRB and p16, are lacking in over 50% of human breast cancers (5), and this was subsequently confirmed by others (15). p16 appears to be the primary target of inactivation. Using an antibody that is quite specific for wild type p16 (14), we confirmed that p16 is absent in almost half of our breast tumors. However, it was evident that in a substantial number of cases, the cell cycle had to be deregulated by other mechanisms. Thus, we probed for overexpression of cyclin D1, the effect of which may, in principle, be comparable to the loss of p16. Likewise, we probed for abnormalities in p53 because this gene also participates in the control of the late G1 restriction point by virtue of its transactivational effect on p21WAF1, a less specific CDK inhibitor (1).
In our series of breast cancers, cyclin D1 was overexpressed in 28 of 81 lesions (35%), which is well within the range of previously reported frequencies (25 to 73%). Using the same antibody and staining cut-off (5%), our data are very similar to those reported by Bartkova et al. (16). Whereas we did not probe for amplification of the gene, it is well documented that gene amplification accounts for less than half of the cases with cyclin D1 overexpression (17, 18, 19). Ductal and lobular carcinomas had similar rates of cyclin D1 overexpression. This is in keeping with previous reports (6, 17, 20, 21, 22), but is at variance with one report of a higher prevalence of cyclin D1 overexpression in lobular versus ductal tumors (23). In our series, an elevated level of cyclin D1 was not associated with important pathological prognostic markers including tumor size, axillary lymph node status, and grade. Most previous reports also failed to demonstrate a correlation between cyclin D1 overexpression and size of the lesion, and no study has yet shown an association with axillary lymph node involvement (6, 18, 19, 20, 21, 22, 23). Likewise, the majority of published series failed to report a correlation with tumor grade (18, 19, 20, 21, 22), although two groups reported a lower cyclin D1 positivity rate in high grade breast cancers (6, 23). It is conceivable that the possible association with low grade may in part explain the favorable prognostic significance of cyclin D1 overexpression in breast cancer reported by some (6). However, the majority of published studies have found no survival difference between cyclin D1 negative and positive tumors. Our data support the concept that persistent elevation of intranuclear cyclin D1 occurs early in mammary neoplasia and is maintained during tumor progression (17). Like two other studies (22, 23), but unlike a third one (20), we found no association between cyclin D1 status and DNA ploidy. We were not able to study the association between positive estrogen receptor status and cyclin D1 overexpression, as has been documented by a number of other investigators (6, 18, 21, 22).
Twenty-five of our 81 breast cancers (31%) overexpressed p53, which is near the average abnormality rate reported by other investigators (range, 11 to 58%). The level of p53 protein in non-neoplastic cells is low but detectable by sensitive immunohistochemical methods. Accumulation of p53 protein is usually interpreted as a surrogate marker for a missense mutation in the gene, which is the most common mechanism of p53 inactivation in breast cancer (9). However, elevated levels of wild type p53 may also result in a positive immunohistochemical reaction, while non-missense mutations are usually not detectable by immunohistochemistry (8, 9). Thus, it is well established that this methodology can only be regarded as a first approximation of p53 dysfunction. Nevertheless, we used immunohistochemistry because of its relative ease and because it allowed us to assess p53 expression in the various compartments of a given tissue. One advantage of the technique is that it enabled us to detect rare cases of intratumoral heterogeneity in p53 expression (Fig. 1B).
Controversy exists whether p53 accumulation is more common in ductal versus lobular carcinomas. The data by Lipponen et al. (24) were affirmative, whereas two other groups found no statistically significant difference (25, 26). In our series, which included a relatively large number of lobular cancers, p53 accumulation occurred at similar rates in both major histological types of breast cancer. Likewise, there was no significant association with tumor size or axillary lymph node status, which is in agreement with the great majority of earlier reports (24, 25, 26, 27, 28, 29, 30, 31, 32). This also suggests that p53 plays an important role in the early stages of mammary neoplasia, but is not responsible for the differences in metastatic potential of established breast cancers. On the other hand, our observation that p53 accumulation is more common in tumors with high mitotic activity and of higher nuclear and overall grade is in keeping with other studies (24, 25, 26, 28, 30) and suggests a role of p53 in the locally aggressive phenotype of primary breast cancers. Whether p53 is important for tumor differentiation remains to be determined. Like Pieteläinen et al. (26), we found no correlation between p53 status and histologic grade, whereas two other groups reported higher p53 accumulation rates in poorly differentiated breast carcinomas (25, 30). Although p53 is thought to be a determinant of genomic instability (33), we and others (24, 26, 27, 31) did not find an association between abnormal p53 expression and DNA aneuploidy. Such a correlation was, however, described by Jacquemier et al. (7).
Whereas numerous publications explored the role of either cyclin D1 or p53 in human mammary neoplasia, we know of only two other immunohistochemical studies on the expression of both proteins in the same cohort of breast tumors. Our findings largely coincide with those of Michalides et al. (21), who studied 243 breast cancers and Barbareschi et al. (22), who reported on 64 tumors. Like both of these groups, we found no correlation between cyclin D1 and p53 abnormalities in the whole cohort. This suggests that the cell cycle regulatory effects of the two genes are not redundant. Alternatively, it is possible that in neoplasms that abrogate the G1 checkpoint by cyclin D1 overexpression, inactivation of p53 is an important independent event that impacts on other cell cycle checkpoints or on the apoptotic pathway. It is noteworthy that high levels of cyclin D1 and p53 were observed in 12% of ductal, but in none of 26 lobular carcinomas. We previously noted the absence of RB inactivation in lobular cancers (5). However, these differences did not reach statistical significance (P = .09 and P = .186, respectively).
We previously described the strict inverse correlation between loss of p16 and inactivation of RB in human mammary neoplasia (5), which has also been noted in many other malignancies. Whereas p53 accumulation was not less common in cyclin D1 overexpressing breast cancers (see above), it was decidedly less frequent in p16 negative tumors (P = .007). This suggests that most mammary neoplasms require only one of these two events to become clinically manifest. It is possible that the deregulatory effect of p16 loss is stronger than that of cyclin D1 upregulation and does not require the additional downregulation of p21WAF1 that may be accompanied by p53 inactivation. Alternatively, the inverse correlation detailed in Table 3 may be related to inactivation of p14ARF, a structurally unrelated tumor suppressor protein that is also encoded by the CDKN2/INK4A locus on chromosome 9p21 (34). Although we did not study the expression of p14ARF, it is reasonable to assume that this protein is missing in many or most of those breast cancers with a homozygous deletion of the p16 gene, which accounts for a high percentage of cases without demonstrable p16 protein expression (35). It has recently been shown that p14ARF acts as a p53 agonist (36). Hence, inactivation of p53 and deletion of CDKN2/INK4A would target the same functional pathway, in which case a mutually exclusive relationship of p53 mutation and loss of p16INK4A/p14ARF in the same neoplasm would be expected. In contrast, loss of p16 and overexpression of cyclin D1 often coexisted in the same tumors, indicating that these two genes can cooperate in the phosphorylation of pRB and, thus, in the abrogation of the G1/S restriction point. In an experimental system, it was shown that loss of p16 and overexpression of cyclin D1 had a synergistic effect on the proliferation of carcinoma cell lines (37). Whether the simultaneous occurrence of both events is associated with a more aggressive clinical phenotype remains to be determined.
Reports of abnormalities in more than two cell cycle regulatory genes in the same group of breast cancers are rare. Barbareschi et al. (22) reported on the expression of pRB, cyclin D1 and p53, but not p16, in 64 breast cancers. Dublin et al. (15) and Nielsen et al. (38) studied breast tumors (n = 192 and n = 114, respectively) for pRB, cyclin D1 and p16, but not p53 expression. Thus, we believe our series of 77 breast cancers to be the first for which immunohistochemical data on all four major cell cycle regulatory proteins (pRB, p16, cyclin D1, and p53) are available. Among the three proteins most closely associated with the late G1 restriction point (pRB, p16, cyclin D1), p16 was most frequently abnormally expressed. Two-thirds of breast cancers in our cohort showed an abnormal expression pattern for at least one of these proteins (Table 4). If p53 was included in the analysis, only 14 of 77 tumors (18%) had a normal expression pattern for all four proteins. Because we would not have detected non-missense mutations in p53 by our immunohistochemical method, this may be a conservative estimate of cell cycle checkpoint deregulation. Interestingly, these frequencies are very similar to those observed by us in a series of 79 invasive bladder carcinomas (4) and provide evidence for the importance of cell cycle deregulation in human neoplasia in general. We did not investigate abnormalities in CDK4, which is also intimately associated with the G1 cell cycle checkpoint. However, An et al. (39) described amplification of this gene in 15 of 95 (16%) breast cancers. It is tempting to speculate that CDK4 amplification may be responsible for cell cycle deregulation in many of the cases that express pRB, p16, cyclin D1, and p53 at normal levels.
We are particularly interested in the biological differences between ductal and lobular carcinomas. Our data show that abnormal expression of cell cycle regulatory genes occurs at comparable frequencies in both types of tumors (Table 4). Given that cell cycle deregulation may be a universal requirement for the establishment of a malignant neoplasm, this observation may not be too surprising.
We hypothesized that tumors in which the cell cycle was abrogated by more than one mechanism may have a more aggressive phenotype. We found a strong correlation between the number of abnormally expressed genes and mitotic activity of the tumor (P = .007), as might be expected. Indeed, carcinomas with mitotic grades of II or III (i.e., more than five mitoses per 10 high power fields) abnormally expressed at least one of the four proteins studied (Table 5). The association with overall tumor grade (P = .035) was largely due to the mitogenic effect of abnormal cell cycle gene expression. Whether tumors with abnormal expression patterns of two or, rarely, three genes are associated with lower survival, remains to be investigated. Somewhat surprisingly, larger or metastatic tumors were not characterized by a larger number of abnormally expressed cell cycle genes compared to smaller or nonmetastatic ones (Table 5). This suggests that the aggressive potential of a breast cancer is determined less by the extent of cell cycle deregulation and more by abnormalities in other pathways. Abrogation of cell cycle control may occur early in the development of a neoplasm, while deregulation of other pathways that occur during tumor progression may determine the metastatic potential of the lesion.
In summary, our study confirms the significant prevalence of p53 and cyclin D1 abnormalities in human breast cancer. Abrogation of the late G1 checkpoint by abnormal expression of one or more of four cell cycle regulatory proteins occurred in some 80% of both ductal and lobular carcinomas. These abnormalities correlated with proliferative activity and grade of the tumors but not with pathologic markers of metastatic potential, suggesting that they are relatively early events in the development of breast cancers.
References
Sherr CJ . Cancer cell cycles. Science 1996; 6: 1672–1677.
Özcelik H, Mousses S, Andrulis IL . Low levels of expression of an inhibitor of cyclin-dependent kinases (CIP1/WAF1) in primary breast carcinomas with p53 mutations. Clin Cancer Res 1995; 1: 907–912.
Geradts J, Fong KM, Zimmerman PV, Maynard R, Minna JD . Correlation of abnormalities of RB, p16ink4a, and p53 expression with 3p loss of heterozygosity, other genetic abnormalities and clinical features in 103 primary non-small cell lung cancers. Clin Cancer Res 1999; 5: 791–800.
Niehans GA, Kratzke RA, Froberg MK, Aeppli DM, Nguyen PL, Geradts J . G1 Checkpoint protein and p53 abnormalities occur in most invasive transitional cell carcinomas of the urinary bladder. Br J Cancer 1999; 80: 1175–1184.
Geradts J, Wilson PA . High frequency of aberrant p16INK4A expression in human breast cancer. Am J Pathol 1996; 149: 15–20.
Gillett C, Smith P, Gregory W, Richards M, Millis R, Peters G . Cyclin D 1 and prognosis in human breast cancer. Int J Cancer 1996; 69: 92–99.
Jacquemier J, Moles JP, Penault-Llorca F, Adélaide J, Torrente M, Viens P, et al. p53 immunohistochemical analysis in breast cancer with four monoclonal antibodies: comparison of staining and PCR-SSCP results. Br J Cancer 1994; 69: 846–852.
Soong R, Robbins PD, Dix BR, Grieu F, Lim B, Knowles S, et al. Concordance between p53 protein overexpression and gene mutation in a large series of common human carcinomas. Hum Pathol 1996; 27: 1050–1055.
Sjögren S, Inganäs M, Norberg T, Lindgren A, Nordgren H, Holmberg L, et al. The p53 gene in breast cancer: prognostic value of complementary DNA sequencing versus immunohistochemistry. J Natl Cancer Inst 1996; 88: 173–182.
Ewers S-B, Attewell R, Baldertorp B, Borg Å, Långström E, Killander D . Prognostic potential of flow cytometric S-phase and ploidy prospectively determined in primary breast carcinomas. Breast Cancer Res Treat 1991; 20: 93–108.
Jacobs TW, Prioleau JE, Stillman IE, Schnitt SJ . Loss of tumor marker-immunostaining intensity on stored paraffin slides of breast cancer. J Natl Cancer Inst 1996; 88: 1054–1059.
Elston CW, Ellis IO . Pathological prognostic factors in breast cancer. The value of histologic grade in breast cancer: experience from a large study with long term follow-up. Histopathology 1991; 19: 403–410.
Geradts J, Kratzke RA, Niehans GA, Lincoln CE . Immunohistochemical detection of the cyclin-dependent kinase inhibitor 2/multiple tumor suppressor gene 1 (CDKN2/MTS1) product p16INK4A in archival human solid tumors: correlation with retinoblastoma protein expression. Cancer Res 1995; 55: 6006–6011.
Geradts J, Hruban R, Schutte M, Kern S, Maynard R . Immunohistochemical p16 INK4a analysis of archival tumors with deletion, hypermethylation, or mutation of the CDKN2/MTS1 gene: a comparison of four commercial antibodies. Appl Immunohistochem Mol Morphol 2000; 8: 71–79.
Dublin EA, Patel NK, Gillett CE, Smith P, Peters G, Barnes DM . Retinoblastoma and p16 proteins in mammary carcinoma: their relationship to cyclin D1 and histological parameters. Int J Cancer 1998; 79: 71–75.
Bartkova J, Lukas J, Strauss M . Cell cycle-related variation and tissue-restricted expression of human cyclin D1 protein. J Pathol 1994; 172: 237–245.
Bartkova J, Lukas J, Müller H, Lützhoft D, Strauss M, Bartek J . Cyclin D 1 protein expression and function in human breast cancer. Int J Cancer 1994; 57: 353–361.
Worsley SD, Jennings BA, Khalil KH, Mole M, Girling AC . Cyclin D 1 amplification and expression in human breast carcinoma: correlation with histological prognostic markers and oestrogen receptor expression. J Clin Pathol: Mol Pathol 1996; 49: M46–M50.
Frierson HF, Gaffey MJ, Zukerberg LR, Arnold A, Williams ME . Immunohistochemical detection and gene amplification of cyclin D1 in mammary infiltrating ductal carcinoma. Mod Pathol 1996; 9: 725–730.
Collecchi P, Passoni A, Rocchetta M, Gnesi E, Baldini E, Bevilacqua G . Cyclin-D1 expression in node-positive (N+) and node-negative (N−) infiltrating human mammary carcinomas. Int J Cancer 1999; 84: 139–144.
Michalides R, Hageman P, van Tinteren H, Houben L, Wientjens E, Klompmaker R, et al. A clinicopathological study on overexpression of cyclin D1 and of p53 in a series of 248 patients with operable breast cancer. Br J Cancer 1996; 73: 728–734.
Barbareschi M, Pelosio P, Caffo O, Buttitta F, Pellegrini S, Barbazza R, et al. Cyclin-D1-gene amplification and expression in breast carcinoma: relation with clinicopathological characteristics and with retinoblastoma gene product, p53 and p21WAF1 immunohistochemical expression. Int J Cancer 1997; 74: 171–174.
van Diest PJ, Michalides RJ, Jannink L, van der Valk P, Peterse HL, de Jong JS, et al. Cyclin D 1 expression in invasive breast cancer. correlations and prognostic value. Am J Pathol 1997; 2: 705–711.
Lipponen P, Ji H, Aaltomaa S, Syrjänen S, Syrjänen K . p53 protein expression in breast cancer as related to histopathological characteristics and prognosis. Int J Cancer 1993; 55: 51–56.
Haerslev T, Jacobsen GK . An immunohistochemical study of p53 with correlations to histopathological parameters, c-erbB-2, proliferating cell nuclear antigen, and prognosis. Hum Pathol 1995; 26: 295–301.
Pietiläinen T, Lipponen P, Aaltomaa S, Eskelinen M, Kosma V-M, Syrjänen K . Expression of p53 protein has no independent prognostic value in breast cancer. J Pathol 1995; 177: 225–232.
Cattoretti G, Rilke F, Andreola S, D’Amato L, Delia D . p53 expression in breast cancer. Int J Cancer 1998; 41: 178–183.
Barbareschi M, Leonardi E, Mauri FA, Serio G, Dalla Palma P . p53, and c-erbB-2 protein expression in breast carcinomas. An immunohistochemical study including correlations with receptor status, proliferation markers, and clinical stage in human breast cancer. Am J Clin Pathol 1992; 98: 408–418.
Bhargava V, Thor A, Deng G, Ljung B-M, Moore DH, Waldman F, et al. The association of p53 immunopositivity with tumor proliferation and other prognostic indicators in breast cancer. Mod Pathol 1994; 7: 361–368.
MacGrogan G, Bonichon F, de Mascarel I, Trojani M, Durand M, Avril A, et al. Prognostic value of p53 in breast invasive ductal carcinoma: an immunohistochemical study on 942 cases. Breast Cancer Res Treat 1995; 36: 71–81.
Cunningham JM, Ingle JN, Jung SH, Cha SS, Wold LE, Farr G, et al. p53 gene expression in node-positive breast cancer: relationship to DNA ploidy and prognosis. J Natl Cancer Inst 1994; 86: 1871–1873.
Seshadri R, Leong AS-Y, McCaul K, Firgaira FA, Setlur V, Horsfall DJ . Relationship between p53 gene abnormalities and other tumour characteristics in breast-cancer prognosis. Int J Cancer 1996; 69: 135–141.
Levine AJ . p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–331.
Haber DA . Splicing into senescence: the curious case of p16 and p19ARF. Cell 1997; 91: 555–558.
Gazzeri S, Gouyer V, Vour’ch C, Brambilla C, Brambilla E . Mechanisms of p16INK4A inactivation in non small-cell lung cancers. Oncogene 1998; 16: 497–504.
Pomerantz J, Schreiber-Agus N, Liégeois NJ, Silverman A, Alland L, Chin L, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998; 92: 713–723.
Lukas J, Aagaard L, Strauss M, Bartek J . Oncogenic aberrations of p16INK4/CDKN2 and cyclin D1 cooperate to deregulate G1 control. Cancer Res 1995; 55: 4818–4823.
Nielsen NH, Emdin SO, Cajander J, Landberg G . Deregulation of cyclin E and D1 in breast cancer is associated with inactivation of the retinoblastoma protein. Oncogene 1997; 14: 295–304.
An H-X, Beckmann MW, Reifenberger G, Bender HG, Niederacher D . Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation. Am J Pathol 1999; 154: 113–118.
Acknowledgements
We thank Rob Maynard for performing the immunohistochemical stains. We are also indebted to Linda Summerville for expert secretarial assistance and to Helena Furberg for performing the statistical analyses.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Geradts, J., Ingram, C. Abnormal Expression of Cell Cycle Regulatory Proteins in Ductal and Lobular Carcinomas of the Breast. Mod Pathol 13, 945–953 (2000). https://doi.org/10.1038/modpathol.3880172
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880172
Keywords
This article is cited by
-
Canine distemper virus induces apoptosis in cervical tumor derived cell lines
Virology Journal (2011)
-
Identification of metastasis-associated breast cancer genes using a high-resolution whole genome profiling approach
Journal of Cancer Research and Clinical Oncology (2011)
-
SCC-112 gene is involved in tumor progression and promotes the cell proliferation in G2/M phase
Journal of Cancer Research and Clinical Oncology (2008)
-
Cyclin D1 protein overexpression and CCND1 amplification in breast carcinomas: an immunohistochemical and chromogenic in situ hybridisation analysis
Modern Pathology (2006)
