
Abstract
Proinflammatory cytokines, including tumor necrosis factor (TNF)α, have been recognized as important physiopathogenetic factors in the initiation and continuation of inflammatory cardiomyopathies. Experimental and preliminary human studies have demonstrated that TNFα plays a crucial role in enteroviral-induced myocarditis. In this study, we investigated the expression of TNFα and both its receptors (TNFRI and TNFRII) in both viral and nonviral myocarditis. Myocardial expression of TNFα was then correlated with different clinical and pathologic findings. TNFα expression was investigated in endomyocardial biopsies obtained from 38 patients with myocarditis and from eight control subjects by using reverse transcriptase-polymerase chain reaction (PCR) and immunohistochemistry. Viral etiology was diagnosed by PCR in 20 cases: enterovirus in seven, Epstein–Barr virus in four, hepatitis C virus in three, adenovirus in two, influenza virus in two, cytomegalovirus in one, and double infection adenovirus and enterovirus in one. Immunohistochemistry was also used to analyze both TNFα receptors (RI and RII). A semiquantitative analysis was employed (score 0–3) for necrosis, inflammation, fibrosis and immunohistochemical findings. TNFα mRNA and TNFα protein were significantly more present in viral myocarditis than in nonviral myocarditis (16/20 vs 3/18, P=0.001). Remarkable immunostaining was observed for both receptors, particularly TNFRI. Histological analysis revealed that myocardial necrosis (mean score 1.89 vs 1.15, P=0.01) and cellular infiltration (mean score 2.26 vs 1.78, P=0.05) were more prominent in TNFα-positive cases. Among TNFα-positive cases, the greater TNFα mRNAs, the more impaired was cardiac function. Our findings suggest that the expression of TNFα may play an important role in the pathogenesis of viral myocarditis of any etiology and may influence the severity of cardiac dysfunction. Cytokine effects are more strictly linked to overexpression of TNFRI.
Similar content being viewed by others
Main
Myocarditis is an inflammation of the myocardium caused by infection and/or autoimmune reactions that may progress to dilated cardiomyopathy and heart failure.1, 2 Several studies, using a molecular approach, have reported that different cardiotropic viruses have been implicated in most cases of human myocarditis and cardiomyopathy.3, 4 Whether myocyte damage in the early phase of the disease is primarily linked to the viral presence or to immunomediated damage is still under discussion; however, it is now accepted that the progression of the disease is mainly sustained by immunomechanisms.5
The presence of heterogeneous inflammatory cell populations in inflammatory cardiomyopathy implies the existence of a complex, cytokine-rich microenvironment that may contribute to the pathogenesis.6, 7
Among cytokines, tumor necrosis factor (TNF)α is thought to play a pivotal role in the development and progression of myocarditis,8, 9, 10, 11, 12 particularly those that are enteroviral related.8, 10 Previous clinical works have demonstrated that there is an association between depressed myocardial function and elevated TNFα mRNA and protein levels either in plasma or in the myocardium of patients (pts) with myocarditis and dilated cardiomyopathy.13, 14
TNFα elicits its biological effects by binding to two distinct cell surface receptors with approximate molecular masses of 55 kDa (TNFRI) and 75 kDa (TNFRII), both expressed in human cardiac myocytes.15, 16, 17 TNFRI has been shown to initiate negative inotropic effects of TNFα, including the cytotoxic effects in adult cardiac myocytes.13
In this study, we investigated the expression of TNFα and its receptor TNFRI and TNFRII in endomyocardial tissues of pts suffering from viral and nonviral myocarditis by using a reverse transcription (RT)-polymerase chain reaction (PCR) method and immunohistochemistry, respectively. Cytokine expression was then correlated with the main clinicohemodynamic and pathologic parameters.
Materials and methods
Patients
A total of 40 endomyocardial biopsies (EMBs) from 38 pts affected by viral and nonviral myocarditis were randomly chosen from pt records in the Cardiac Units of Padua and Rome University Hospitals in the period 1999–2000. Pts included 27 males and 11 females with a mean age of 36±15 years (range 15–76 years). Myocarditis was diagnosed on the basis of clinical symptoms (eg upper respiratory illness, fever, chest pain, arrhythmias, murmurs, cardiomegaly, elevated erythrocyte sedimentation rate, leukocytes, conduction disturbances) in conjunction with histologic signs of inflammatory infiltrate and myocyte damage, according to the Dallas criteria.18, 19 Pts were excluded if there was any angiographic evidence of coronary heart disease, ischemic changes during exercise testing, systemic hypertension, concomitant systemic or endocrine disease that might cause impairment of left ventricular function, or excessive alcohol consumption. Specific types of myocarditis, such as rheumatic, septic, mycotic, eosinophilic, giant cell types, and myocarditis associated with collagen disease and sarcoidosis, were not considered in this study.
A full clinical assessment was performed in all pts, including familiar and personal history of preceding viral illnesses, period of the infection (season), time between onset of symptoms and EMB, and complete physical examination with functional classification according to the New York Heart Association (NYHA). All pts were followed up at 4-week to 3-month intervals. At each visit, clinical assessment, ECG, and 2D echocardiography were performed (Table 1).
Myocardial biopsies from eight donor hearts before transplantation were added as controls (mean age of 47.8 years; range 33–56 years).
Right ventricular EMBs tissues were obtained from all pts by the standard percutaneous right femoral approach using a Cordis bioptome.20 In all, 4–5 tissue samples were retrieved from each pt. Four of these were fixed in 10% buffered formalin and embedded in paraffin after dehydration in a graded alcohol series. The remaining sample was frozen immediately after biopsy at −80°C and used for molecular analysis. In four pts, one heart transplanted (case 1) and three autoptic cases (cases 11, 27, 36), the whole heart has been also evaluated.
All invasive cardiac examinations were approved by the Ethical Committee of both Institutions and performed after informed consent.
Morphological Analyses and Viral Detection
For histology, 4-μm thick sections were cut and stained with hematoxylin and eosin, Weigert van Gieson's, and Heidenhein trichrome and examined by light microscopy.
Immunohistochemistry for the characterization of inflammatory infiltrate was carried out by use of the following antibodies (all Dako Glostrup, Denmark): CD45 (1:20), CD43 (1:40), CD45RO (1:100), CD20 (1:100), CD3 (1:50), CD68 (1:50), CD4 (1:100), and CD8 (1:100). The immunoreaction products were developed using the avidin–biotin–peroxidase complex method.21
In each biopsy, necrosis, the intensity of the infiltrate as well as the extension of fibrosis were graded semiquantitatively as follows:22 score 0=absent; score 1=mild; score 2=moderate; score 3=severe.
For viral detection, total RNA and DNA were extracted from EMBs of pts and control subjects by using the modified RNAzol method.23 The oligonucleotides used to ascertain the quality of extracted RNA or DNA were complementary to the mRNA glyceraldehyde-3-phosphate dehydrogenase (3GPDH) and β-globin gene, respectively.24, 25 PCR analysis was repeated in all negative viral amplicons by using paraffin-embedded samples in order to exclude false-negative due to only one frozen processed sample. As for the paraffin-embedded biopsies, the dried tissue pellets obtained after deparaffinizzation with xylene, were digested in two different lysis solutions for RNA26 and DNA.27 PCR/RT-PCR or nested-PCR and sequencing analysis were used to detect and characterize well-known cardiotropic viruses. Direct cycle sequencing of PCR products was carried out on an ABI model 310 DNA sequencer as previously described.4 Sequence data were analyzed using the sequence analysis 2.1.2 and sequence navigator for sequence comparison.
Blast search was used to identify homology between sequences and all published DNA reported in the Gene Bank Data Base.
Intramyocardial TNFα
TNFα production (TNFα mRNA and TNFα protein) was investigated in each case of myocarditis and in controls.
Myocardial TNFa gene expression
TNFα expression was investigated by RT-PCR. The following oligonucleotide primer pairs were used: sense 5′-CAGAGGGAAGAGTTCCCCAG-3′, antisense 5′-CCTTGGTCTGGTAG GGAGA CG-3′ (325 bp).28
The cycle number (38 cycles) was chosen to ensure that the amount of products synthesized was proportional to the amount of specific mRNA in the original preparation.
Following PCR amplification, PCR products (15 μl) were subjected to electrophoresis on 2% agarose gels containing 0.03 μg/ml ethidium bromide. Among TNFα-positive pts, TNFα mRNA was assessed in 11 cases (in which adequate quantity of RNA was available) with different cardiac dysfunction: left ventricular ejection fraction (EF) <40% (NYHA≥3) in cases 1, 10, 11, 16, 28, and 32 vs EF≥40% (NYHA<3) in cases 19, 24, 26, 29, and 36. The quantification of transcript level was carried out by scanning photographs of gels and analyzing the area under peaks, using Quantity one Biorad software. Levels of mRNA expression were normalized by calculating them as a percentage of 3GPDH mRNA expression levels.29 The band intensity for 3GPDH did not differ significantly within experiments.
Myocardial TNFα protein
After retrieving antigens from a microwave oven and blocking endogenous peroxidase activity with methanol and hydrogen peroxide, the sections were first blocked for 30 min at room temperature with goat serum and then incubated with primary antibody (polyclonal for TNFα 1:250 dilution Genzyme Corp., Cambridge, USA) in a humidified chamber overnight at 4°C. Control slides were stained with secondary antibody alone (1:250 dilution). The slides were then rinsed in PBS five times and incubated for 30 min at room temperature with a biotin-conjugated goat anti-rabbit secondary antibody. The slides were stained with an avidin–biotin complex by use of a peroxidase reporter (Vectastain ABC Kit, Vector Peterborough, UK). Diaminobenzidine was used as the chromogen to visualize the presence and distribution of the cytokine. Quality control sections were obtained after incubation with nonimmune isotype immunoglobulin. Omission of the primary antibody served as a negative control. A positive immune reaction was indicated by a diffuse brown color of the cytoplasm; the expression of the antigens was evaluated independently by two investigators. Immunoassaying was evaluated semiquantitatively as previously described.30
In particular, the extent of immunoreactivity was semiquantitatively scored as follows: 0=absence of immunoreactive cells; score 1=1–10% immunoreactive cells, score 2=11–50% immunoreactive cells, score 3= >50% immunoreactive cells.
TNFa receptor (RII and RII) protein
Serial sections were incubated with primary antibodies (monoclonal for TNFRI and RII both with a 1:250 dilution Genzyme Corp.) for 1 h and with secondary antibody for 10 min. The avidin–biotin complex incubation for 10 min and 3,3-diaminobenzidine were used for visualization. The same semiquantitative analysis (score 0–3) was used.
Statistical Analysis
Results are expressed as mean±s.e.m. One-way analysis of variance (ANOVA) and the Student's t-test for unpaired data were used for differences between subgroup. Selected variables were compared by linear regression analysis. The difference was considered statistically significant at a level of P<0.05. Morphological and immunohistochemical values were analyzed by two pathologists (FC and AA). Inter- and intraobserver variability was determined by κ-statistic method. Pair data were then analyzed using the weighted k coefficient (κ) to determine the level of agreement between the two pathologists. The following scores indicate the relative agreement for values of the κ score defined according to the criteria of Landis and Koch:31 >0.8=almost perfect agreement; 0.6–0.8= substantial agreement; 0.4–0.6=moderate agreement; 0.2–0.4=fair agreement; 0.0–0.2= slight agreement; 0.0= poor agreement.
Results
Clinicohemodynamic Features of Patient Cohort
The majority of pts (90%) showed symptoms lasting less than 24 weeks. In five pts (13%), a flu-like syndrome occurred at least 2 weeks before the onset of cardiac dysfunction, mainly in autumn and winter (23/38; 61%). A familiar history was reported in only two pts (5%). All pts had regional or global left ventricular dysfunction as assessed by echocardiography. An overview of the clinical data at baseline and follow-up is given in Table 1. Adverse outcome occurred in 12 pts (7 deaths and 5 transplants).
Morphological and Viral Findings
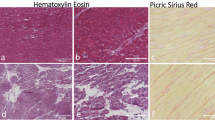
Myocarditis was active in 33 and borderline in five cases (Figure 1a). Immunophenotypical characterization of inflammatory cells showed a prevalence of T lymphocytes (CD45RO and CD3) (Figure 1b), including a moderate amount of cytotoxic lymphocytes (CD8 positive).

Active myocarditis (case 29; EV PCR positive). (a) Severe and diffuse inflammatory cell infiltration with evident necrosis of adjacent myocytes (original magnification × 40). (b) Lymphocytes show strong CD45RO immunoreactivity (original magnification × 40).
In all cases, the presence of sufficient target nucleic acid for PCR analysis was confirmed by amplification of β-globin for DNA and 3GPDH for RNA. Viral genomes were present in 20 pts including seven enteroviruses (35%), two adenoviruses (10%), four Epstein-Barr viruses (20%), two influenza viruses (10%), three hepatitis C viruses (15%), one cytomegalovirus (5%) and one double infection adenovirus–enterovirus (5%) (Table 2). Sequencing analysis of the PCR products showed a high homology (>95%) with published human viral genomes.
None of the control samples showed positive amplification for any cardiotropic viruses investigated in the study pts.
Myocardial TNFα and TNFα Receptors (RI and RII)
TNFα mRNA was detected in 19 out of 38 cases (50%) (Table 2). There was no evidence of TNFα mRNA expression in the controls.
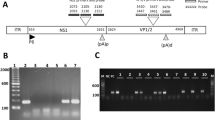
TNFα gene expression was seen in 16 out of 20 viral positive myocarditis cases (80%) vs only three out of 18 nonviral cases (17%) (P=0.001) (Figure 2a, b).

(a) Agarose gel electrophoresis for RT-PCR and PCR of RNA virus and DNA virus with correspondent mRNA TNFα expression. Lane 1: EMB positive for enterovirus case 11 (297 bp); lane 2: TNFα expression of case 11 (325 bp); lane 3: EMB positive for cytomegalovirus case 27 (280 bp); lane 4: TNFα expression of case 27; lane 5: EMB positive for hepatitis C virus case 2 (256 bp); lane 6: TNFα expression of case 2; lane 7: EMB positive for influenza B virus case 19 (241 bp); lane 8: TNFα expression of case 19; lane 9: EMB positive for influenza A virus case n. 10 (190 bp); lane 10: TNFα expression of case 10; lane 11: negative control; lane 12: Marker VIII; lane 13: EMB positive for adenovirus case 6 (308 bp); lane 14: TNFα expression of case 6; lane 15: EMB positive for Epstein–Barr virus case 13 (268 bp); lane 16: TNFα expression of case 13. (b) TNFα expression in viral and in nonviral myocarditis. Cytokine mRNA was detected in 16 out of 20 viral positive cases vs only three out of 18 nonviral cases (P=0.001). No evidence for TNFα mRNA expression in the controls.
TNFα immunostaining showed a marked positivity mainly in the myocardium obtained from viral cases (mean 1.85±0.2 vs 0.6±0.2, P=0.002) and was detected in the cytoplasm of cardiac myocytes other than macrophages and mononuclear cells (Figure 3).

Immunohistochemical analysis for TNFα in endomyocardial biopsy tissue obtained from patient with active myocarditis (case 10). Marked TNFα immunoreactivity is seen in myocytes. Positive cytoplasmic staining is also visible in infiltrating macrophages and mononuclear cells. Original magnification × 60.
Immunohistochemistry for TNFαRI and RII showed that both were overexpressed in TNFα-positive cases, particularly TNFαRI (P=0.001) (Table 2) (Figures 4a, b and 5).

Immunohistochemical analysis for TNF RI and RII: strong (a) and mild (b) staining for TNFRI TNFRII in the same EMB (case 11). Original magnification × 60.

Effect of TNFα expression on TNF RI and RII. As shown, degree of TNFRI is linearly related to the expression of TNFα protein (P=0.001) (R-valueTNFalfa/TNFRI=0.93; R-valueTNFalfa/TNFRII=0.50).
A second follow-up EMBs was performed in two pts (cases 2 and 25); the expression of cytokine and its receptors showed similar results as the previous biopsy.
The large specimens taken from the whole heart of cases 1, 11, 27, and 36, showed the same expression of TNFα pathway as seen in the EMB of the same pts.
In the controls, TNFα immunostaining was always negative, while weak positivity was observed for TNFRI and RII.
There was a full concordance between the two pathologists: κ-score for TNFα immunostaining was 0.88 and TNFRI and RII immunostaining 0.90 and 0.82, respectively.
Correlation between TNFα Expression and Pathological/Clinical Findings
Diffuse myocyte damage was observed more frequently in TNFα-positive myocarditis (score 1.89 vs 1.15, P=0.01). Only a trend for more severe inflammatory infiltrate was noted in TNFα-positive cases (score 2.26 vs 1.78, P=0.05, NS). Instead, no difference in terms of extension of fibrosis was noted between TNFα-positive and negative cases (P=1, NS).
No statistically significant correlation was found between TNFα PCR-positive and TNFα PCR-negative pts compared to various clinical parameters (familiar and personal history of preceding viral illnesses, season, time between onset of symptoms and EMB, EF, LVEDV, and NYHA). However, among TNFα-positive cases, TNFα mRNAs were greater in pts with more impaired ejection fraction. Densitometric analysis revealed that the mRNA levels for TNFα were significantly increased in cases with more depressed EF compared to cases with less EF (3.20±0.8 vs 0.22±0.03, P=0.003) (Figure 6).

Quantification of transcript level of TNFα mRNA expression. Data shown are the mean±s.e.m. of six pts with more impaired function vs five pts with mild impaired function level (P=0.003). EF: left ventricular ejection fraction.
No difference was observed between TNFα-positive and -negative in terms of survival (Table 1). Conversely, an improvement of cardiac function was reported in TNFα-negative pts (NYHA class from 1.3 to 2.5, P=0.01).
Discussion
In this study, we have demonstrated that TNFα mRNA and protein are overexpressed in pts with viral myocarditis of any etiology.
Several studies have suggested that proinflammatory cytokines, such as TNFα, play an important role in the pathogenesis of myocarditis. Increased circulating TNFα has been reported in pts with myocarditis and cardiomyopathy.9 The possible adverse impact of cytokines in myocarditis may cause an altered susceptibility to the disease thus impairing the clinical course. Lane et al32 showed that mice resistant to coxsackievirus infection develop inflammatory lesions when treated with TNFα or IL1. Similar results were found by Yamada et al,33 who also showed that the treatment of mice with monoclonal anti-TNFα antibody before viral inoculation led to reduced mortality of infected animals. Recently, some authors have shown that cytokines upregulate coxsackie–adenovirus receptor (CAR) on human myocardium and that the level of CAR upregulation is a major determinant of susceptibility to cardiac infection by adenoviruses and coxsackie viruses.34
Although the exact mechanism through which TNFα contributes to decreased contractile performance in myocarditis is not well known, a number of studies have emphasized the negative impact mediated by nitric oxide (NO). Finkel et al35 showed that the depression of isolated papillary muscle contractile function caused by TNFα or IL6 was blocked when an L-arginine analogue was added as a specific inhibitor of NO synthase (NOS). Joe et al36 demonstrated that shortening of myocytes is impaired when they are exposed to media in which macrophages are stimulated with lipopolysaccharide and that this can be blocked by inhibition of NOS.
TNFα, produced by activated macrophages and endothelial cells, can cause an increase of inducible NOS activity in cardiac myocytes, and can modulate inotropic responsiveness.37 Other studies have suggested that inflammatory cytokines, including TNFα, may also cause a direct negative inotropic response.38 Yokoyama et al39 have shown that TNFα induces a direct negative inotropic effect in isolated ventricular myocytes of adult cat that is associated with a decrease in the Ca2+ transit, without changing the current L-type Ca2+, and that it is not inhibited by blockers of NO production or arachidonic acid metabolism.
Although the majority of studies emphasize the negative influence of TNFα in different cardiovascular diseases, its role in the setting of myocarditis is still controversial. The amount and the time of endogenous TNFα released in cases of myocarditis seem today to be crucial for the beneficial and adverse effects of the cytokine.40 At low concentrations, TNFα regulates paracrine and autocrine activity of leukocytes and endothelial cells, thus becoming an important monitor of the inflammatory response. TNFα enhances chemotaxis of macrophages and neutrophils, increases their phagocytic and cytotoxic activity41 and promotes leukostasis by inducing increased expression of intracellular adhesion molecules and endothelial leukocyte adhesion molecules at sites of inflammation. Therefore, the presence of TNFα, as recently described by Wada et al,42 plays a protective role in the early stage of encephalomyocarditis virus myocarditis, possibly through leukocyte recruitment, thus hindering viral replication. On the contrary at higher concentrations, TNFα production exceeds the number of its receptors located on the cell surface with an excess amount released into circulation. Once released, it produces devastating consequences not only in terms of multiorgan dysfunction but overall in the setting of cardiovascular system.43, 44
One of the most important findings of this study was a significantly elevated level of expression of TNFα mRNAs and proteins in pts affected by viral myocarditis with lower EF and more impaired NYHA functional class.
The persistent myocardial overexpression of cytokine, as demonstrated in the follow-up biopsy or in the whole heart from pts who died or underwent heart transplantation, is an additional data suggesting that the cytokine is implicated in the progression of the disease and in the development of irreversible heart failure. In fact, an improvement of cardiac function was observed in none of our pts with TNFα overexpression.
Among viral myocarditis cases, the role of TNFα has been evaluated only in enteroviral myocarditis, both in experimental studies32, 45 and human forms,9 and recently also in a few pts with parvovirus B19 myocarditis.46 In our study, we demonstrated that TNFα was significantly more expressed in viral myocarditis than in nonviral forms (16/20 vs 3/18, P=0.01), regardless of the viral etiology: capside or viral proteins have been demonstrated to be responsible for the attraction mainly of Th1 cells with consequently over-release of specific proinflammatory cytokines, just including TNFα.
Concerning the cell origin of TNFα, activated macrophages as well as lymphocytes and endothelial cells are considered the principal source in the course of myocarditis.39 Several studies have demonstrated that in different cardiac-related pathologic conditions (congestive heart failure, myocarditis, dilated cardiomyopathy, ischemic heart disease, and septic cardiomyopathy) cardiac myocytes are actively involved in cytokine biosynthesis.13, 47 Our immunohistochemical analysis confirmed that the cellular source of TNFα protein was not only macrophages and mononuclear cells but also the cardiac myocytes themselves. Thus, TNFα produced by myocytes might have an autocrine effect, thus contributing to the progression of myocyte damage.
The direct intramyocardial evaluation of TNFα, with molecular techniques and immunohistochemistry, appears undoubtedly more satisfactory than circulating levels which may not reflect the specific targeted organs.48 Furthermore, in congestive heart failure some authors have demonstrated that there is no significant correlation between the level of TNFα in the myocardium and the levels in plasma, probably because of its multiple sites of production.39 Consequently, TNFα expression in our study was restricted to intramyocardial cytokine detection. A significant more prominent myocardial damage and well-evident inflammation was observed in TNFα-positive than TNFα-negative myocarditis. The severe myocyte damage may be either caused by TNFα directly and by inflammatory cells that are recruited by TNFα, by the expression of other cytokines, or by the induction of NOS and the generation of free radicals.
A higher level of TNFα expression was recently reported in biopsies from pts affected by myocarditis with more extensive necrosis and inflammation; the authors noted a marked decrease of TNFα expression when cellular infiltration had become reduced.49 However, viral etiology of myocarditis was not investigated at that time.
In our clinical study, we have also demonstrated an overexpression of TNFRI. Several experimental studies have shown the potential importance of TNFRI in signaling the negative inotropic effects of TNFα. TNFRI-deficient transgenic mice are resistant to the toxic effects of systemic TNF-α as well as to the systemic toxicity of lipopolysaccharide or Staphylococcus aureus enterotoxin B.50, 51 On the other hand, other comparable studies on TNFRII-deficient mice showed that even though the mice were less sensitive to the toxic effects of systemic TNFα, they were not completely resistant to the adverse effects of TNFα as were the TNFRI-deficient mice.52
The finding of intramyocardial TNFα in viral myocarditis provides not only new insights to the pathogenesis of viral forms but also suggests new targets for therapeutic intervention. Two large randomized clinical trials in human heart failure, aimed at determining the effect, of potent anti-TNFα-blocking agents (etanercept and infliximab) on mortality, failed to demonstrate clinical benefit.53 New approaches should be planned in the future focusing on myocarditis and intramyocardial TNFα overexpression.
References
Billingham ME, Tazelaar HD . The morphological progression of viral myocarditis. Postgrad Med J 1986;62:581–584.
O'Connell JB . The role of myocarditis in end-stage dilated cardiomyopathy. Texas Heart Inst J 1987;14:268–275.
Martin AB, Webber S, Fricker FJ, et al. Acute myocarditis. Rapid diagnosis by PCR in children. Circulation 1994;90:330–339.
Calabrese F, Rigo E, Milanesi O, et al. Molecular diagnosis of myocarditis and dilated cardiomyopathy in children: clinicopathologic features and prognostic implications. Diagn Mol Pathol 2002;11:212–221.
Liu PP, Mason JW . Advances in the understanding of myocarditis. Circulation 2001;104:1076–1082.
Neumann DA, Lane JR, Allen GS, et al. Viral myocarditis leading to cardiomyopathy: do cytokines contribute to pathogenesis? Clin Immunol Immunopathol 1993;68:181–190.
Gluck B, Schmidtke M, Merkle I, et al. Persistent expression of cytokines in the chronic stage of CVB3-induced myocarditis in NMRI mice. J Mol Cell Cardiol 2001;33:1615–1626.
Gillespie JS, Cavanagh HM, Behan WM, et al. Increased transcription of interleukin-6 in the brains of mice with chronic enterovirus infection. J Gen Virol 1993;74:741–743.
Matsumori A, Yamada T, Suzuki H, et al. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br Heart J 1994;72:561–566.
Satoh M, Tamura G, Segawa I, et al. Expression of cytokine genes and presence of enteroviral genomic RNA in endomyocardial biopsy tissues of myocarditis and dilated cardiomyopathy. Virchows Arch 1996;427:503–509.
Matsumori A . Cytokines in myocarditis and cardiomyopathies. Curr Opin Cardiol 1996;11:302–309.
Kelleher Doyle M, Telford III SR, Criscione L, et al. Cytokines in murine lyme carditis: Th1 cytokine expression follows expression of proinflammatory cytokines in a susceptible mouse strain. J Infect Dis 1998;177:242–246.
Torre-Amione G, Kapadia S, Lee J, et al. Tumor necrosis factor-α and receptor in the failing heart. Circulation 1996;93:704–711.
Habib FM, Springall DR, Davies GJ, et al. Tumor necrosis factor and inducible nitric oxide synthase in dilated cardiomyopathy. Lancet 1996;347:1151–1155.
Dembic Z, Loetscher H, Gubler U, et al. Two human TNF receptors have similar extra cellular, but distinct intracellular, domain sequences. Cytokine 1990;2:231–237.
Vassalli P . The pathophysiology of tumor necrosis factors. Annu Rev Immunol 1992;10:411–452.
Beutler B, van Huffel C . Unraveling function in the TNF ligand and receptor families. Science 1994;264:667–668.
Aretz HT, Billingham ME, Edwards WD, et al. Myocarditis. A histopathologic definition and classification. Am J Cardiovasc Pathol 1987;1:3–14.
Aretz HT . Myocarditis: the Dallas criteria. Hum Pathol 1987;18:619–624.
Richardson PJ . King's endomyocardial bioptome. Lancet 1974;18:660–664.
Cartun RW, Pedersen CA . An immunocytochemical technique offering increased sensitivity and lowered cost with a streptavidin–horseradish peroxidase conjugate. J Histotechnol 1989;12:273–277.
Angelini A, Crosato M, Boffa GM, et al. Active versus borderline myocarditis: clinicopathological correlates and prognostic implications. Heart 2002;87:210–215.
Chomczynski P, Sacchi N . Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem 1987;162:156–159.
Ercolani L, Florence B, Denaro M, et al. Isolation and complete sequence of a functional human glyceraldehyde-3-phosphate dehydrogenase gene. J Biol Chem 1988;263:1535–1541.
Saiki RK, Scharf S, Faloona F, et al. Enzymatic amplification of betaglobin genomic sequence and restriction site analysis for diagnosis of sickle cell anemia. Science 1985;230:1350–1354.
Stanta G, Schneider C . RNA extracted from paraffin-embedded human tissues is amenable to analysis by PCR amplification. Biotechniques 1991;11:304–308.
Shibata DK . The polymerase chain reaction and the molecular genetic analysis of tissue biopsies. In: Rickwood D, Hames BD (eds). Diagnostic Molecular Pathology, A practical Approach. Vol II. Oxford Edition by Herrington CS, McGee O'D. 1992, pp 85–110.
Yamaguchi S, Kitagawa M, Inoue M, et al. Cell dynamics and expression of tumor necrosis factor (TNF)-α, interleukin-6 and TNF receptors in angioimmunoblastic lymphadenopathy-type T cell lymphoma. Exp Mol Pathol 2000;68:85–94.
Horikoshi T, Sakakibara M . Quantification of relative mRNA expression in the rat brain using simple RT-PCR and ethidium bromide staining. J Neurosci Methods 2000;99:45–51.
Barrera P, Joosten LA, den Broeder AA, et al. Effects of treatment with a fully human anti-tumour necrosis factor alpha monoclonal antibody on the local and systemic homeostasis of interleukin 1 and TNFalpha in patients with rheumatoid arthritis. Ann Rheum Dis 2001;60:660–669.
Landis JR, Koch GG . An application of hierarchical kappa-type statistics in the assessment of majority agreement among multiple observers. Biometrics 1977;33:363–374.
Lane JR, Neumann DA, Lafond-Walker A, et al. Interleukin-1 or tumor necrosis factor can promote coxasackie B3- induced myocarditis in resistant B10. J Exp Med 1992;175:1123–1129.
Yamada T, Matsumori A, Saayama S . Therapeutic effect of anti-tumor necrosis factor-α antibody on the murine model of viral myocarditis induced by encephalomyocarditis virus. Circulation 1994;89:846–851.
Noutsias M, Fechner H, de Jonge H, et al. Human coxsackie–adenovirus receptor is colocalized with integrins αvβ3 and αvβ5 on the cardiomyocyte sarcolemma and upregulated in dilated cardiomyopathy. Implications for cardiotropic viral infections. Circulation 2001;104:275–280.
Finkel MS, Oddis CV, Jacob TD, et al. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 1992;257:387–389.
Joe EK, Schussheim AE, Longrois D, et al. Regulation of cardiac myocyte contractile function by inducible nitric oxide synthase (iNOS): mechanisms of contractile depression by nitric oxide. J Mol Cell Cardiol 1998;30:303–315.
Gulick T, Pieper SJ, Murphy MA, et al. A new method for assessment of cultured cardiac myocyte contractility detects immune factor-mediated inhibition of beta-adrenergic responses. Circulation 1991;84:313–321.
Barry WH . Mechanisms of immune mediated myocyte injury. Circulation 1994;89:2421–2432.
Yokoyama T, Vaca L, Rossen RD, et al. Cellular basis for the negative inotropic effects of tumor necrosis factor-α in the adult mammalian heart. J Clin Invest 1993;92:2303–2312.
Feldman AM, Combes A, Wagner D, et al. The role of tumor necrosis factor in the pathophysiology of heart failure. J Am Coll Cardiol 2000;35:537–544.
Djeu JY, Blanchard DK, Richards AL, et al. Tumor necrosis factor induction by Candida albicans from human natural killer cells and monocytes. J Immunol 1988;141:4047–4052.
Wada H, Saito K, Kanda T, et al. Tumor necrosis factor-α plays a protective role in acute viral myocarditis in mice. A study using mice lacking TNFα. Circulation 2001;103:743–749.
Vilcek J, Lee TH . Tumor necrosis factor. J Biol Chem 1991;266:7313–7316.
Levine B, Kalman J, Mayer L, et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 1990;223:236–241.
Shioi T, Matsumori A, Sasayama S . Persistent expression of cytokine in the chronic stage of viral myocarditis in mice. Circulation 1996;94:2930–2937.
Nigro G, Bastianon V, Colloridi V, et al. Human parvovirus B19 infection in infancy associated with acute and chronic lymphocytic myocarditis and high cytokine levels: report of 3 cases and review. Clin Infect Dis 2000;31:65–69.
Jibiki T, Terai M, Tateno S, et al. Expression of tumor necrosis factor-alpha protein in the myocardium in fatal myocarditis. Pediatr Int 2000;42:43–47.
Bryant D, Becker L, Richardson J, et al. Cardiac Failure in transgenic mice with myocardial expression of tumor necrosis factor-α. Circulation 1998;97:1375–1381.
Satoh M, Nakamura M, Satoh H, et al. Expression of tumor necrosis factor-alpha-converting enzyme and tumor necrosis factor-alpha in human myocarditis. J Am Coll Cardiol 2000;36:1288–1294.
Jibiki T, Terai M, Tateno S, et al. Mice lacking the tumor necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993;3:798–802.
Pfeffer K, Matsuyama T, Kundig TM, et al. Mice deficient for the 55 kD tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993;73:457–467.
Erickson SL, de Sauvage FJ, Kikly K, et al. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature 1994;372:560–563.
Louis A, Cleland JG, Crabbe S, et al. Clinical trials update: CAPRICORN, COPERNICUS, MIRACLE, STAF, RITZ-2, RECOVER and RENAISSANCE and cachexia and cholesterol in heart failure. Highlights of the Scientific Sessions of the American College of Cardiology, 2001. Eur J Heart Fail 2001;3:381–387.
Acknowledgements
This work was supported by Grant Project ‘Myocarditis: therapeutic impact of etiological diagnosis based upon molecular and immunologic findings’, Ministery of Education, University and Research, Rome, Italy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Calabrese, F., Carturan, E., Chimenti, C. et al. Overexpression of tumor necrosis factor (TNF)α and TNFα receptor I in human viral myocarditis: clinicopathologic correlations. Mod Pathol 17, 1108–1118 (2004). https://doi.org/10.1038/modpathol.3800158
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800158
Keywords
This article is cited by
-
The viral distribution and pathological characteristics of BALB/c mice infected with highly pathogenic Influenza H7N9 virus
Virology Journal (2021)
-
Myopericarditis and exertional rhabdomyolysis following an influenza A (H3N2) infection
BMC Infectious Diseases (2013)
-
Diagnostic use of the endomyocardial biopsy: a consensus statement
Virchows Archiv (2013)
-
Classification and histological, immunohistochemical, and molecular diagnosis of inflammatory myocardial disease
Heart Failure Reviews (2013)
-
The expression and significance of protooncogene c-fos in viral myocarditis
Virology Journal (2010)
