
Abstract
Fas ligand (FasL), an apoptosis-inducing cytokine, is constitutively expressed on endothelial cells (EC). Here, we report that the soluble form of FasL (sFasL) is released from EC and inhibits hypoxia-induced EC apoptosis. For hypoxia experiments, human EC were exposed to low oxygen tension in airtight chambers flushed with preanalyzed gas mixtures (1% oxygen, 5% CO2, 94% N2) at 37° C. Exposure of cultured EC to hypoxia transiently increased FasL mRNA and protein levels. The maximum increase was observed at 3 and 6 hours after exposure to hypoxia, respectively. Although sFasL protein was not detected in the supernatant from EC without hypoxia, sFasL protein level in the supernatant was transiently increased from 6 hours and disappeared again at 24 hours after the exposure to hypoxia. Interestingly, the supernatant from hypoxia-exposed EC inhibited EC apoptosis induced by hypoxia, which was abolished by a neutralizing antibody against FasL. In addition, incubation with KB8301, an inhibitor of metalloproteinase, suppressed the release of sFasL from EC and enhanced hypoxia-induced apoptosis in EC. Furthermore, exogenously added recombinant sFasL inhibited hypoxia-induced apoptosis. These findings indicate that sFasL released from EC may inhibit hypoxia-induced EC apoptosis. Therefore, the shedding of FasL could be a new therapeutic target in regulating hypoxia-induced EC injury.
Similar content being viewed by others

Introduction
Apoptosis is an important physiologic cell death mechanism to eliminate damaged or unwanted cells, whereas dysregulated apoptosis has been implicated in many human diseases such as cancer, neurodegenerative diseases, and ischemic heart disease (Thompson, 1995). Recent evidence has shown that atherogenic factors such as oxidized low-density lipoprotein (Sata and Walsh, 1998a), angiotensin II (Li et al, 1999), oxidative stress, inflammatory cytokines (Dimmeler et al, 1998), and hypoxia (Stempien-Otero et al, 1999) induce endothelial cell (EC) apoptosis, whereas antiatherogenic factors such as estrogen (Spyridopoulos et al, 1997) and high-density lipoprotein (Suc et al, 1997) inhibit EC apoptosis. Additionally, Bombeli et al (1997, 1999) reported that apoptotic EC become procoagulant and proadhesive to nonactivated platelets, which suggests that EC apoptosis may be involved in the pathogenesis of atherosclerosis.
Because deprivation of oxygen (hypoxia) is a critical stress for mammalian cells, there are several adaptive responses to restore oxygen homeostasis. Homeostatic responses elicited by hypoxia include erythropoiesis, neovascularization, and glycolysis. Hypoxia-inducible factor 1α, a transacting factor, has been shown to play important roles in activating these adaptive responses (Iyer et al, 1998; Semenza et al, 1994; Wenger and Gassmann, 1997). Recently, Stempien-Otero et al (1999) reported that EC are resistant to hypoxia-induced apoptosis, requiring a prolonged period of exposure to undergo apoptosis, which suggests the presence of intrinsic mechanisms in EC that protect them from hypoxia-induced apoptosis. However, the precise mechanisms of EC survival under hypoxia are not clear.
Fas ligand (FasL; also called CD95 ligand or APO-1 ligand) is a cytokine that mediates apoptosis by binding to its receptor, Fas (also called CD95 or APO-1) (Krammer et al, 1994; Nagata, 1997). FasL is mainly expressed on NK cells, activated T cells, and immune-privileged tissues of the eye and testis, as well as by some tumors where it may contribute to their ability to evade immune detection (Griffith et al, 1995; Hahne et al, 1996; Strand et al, 1996). Recently, Sata and Walsh (1998b) reported that EC constitutively express FasL and that they may have a function to inhibit leukocyte extravasation through inducing apoptosis in adherent mononuclear cells. FasL, a member of the TNF family, is rapidly cleaved off from the membrane by a metalloproteinase to become a soluble form (sFasL) as well as TNFα (Tanaka et al, 1996). Tanaka et al (1998) recently reported that sFasL inhibits Fas-mediated lymphocyte apoptosis. Thus, we examined whether sFasL can modulate hypoxia-induced EC apoptosis. We found that hypoxia stimulated sFasL release from EC, which then protected EC from hypoxia-induced apoptosis.
Results
Hypoxia Up-Regulates FasL Expression
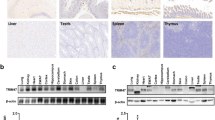
Figure 1 shows the immunocytochemical staining of FasL expression in EC. FasL was constitutively expressed in EC, as previously reported (Sata and Walsh, 1998b). Hypoxia induced up-regulation of FasL expression in EC. To measure the level of FasL expression in EC, we next performed Western blot analysis of FasL protein in EC. The protein level of cellular FasL was transiently increased from 3 hours, reached a peak at 6 hours, and returned to the basal level at 24 hours after exposure to hypoxia (Fig. 2A). Flow cytometric analysis with different antibodies to FasL also showed that the level of cell surface membrane FasL (mFasL) was significantly increased after hypoxic stimulation (Fig. 2B). This up-regulation was enhanced in the presence of KB8301, an inhibitor of metalloproteinase. To define the mechanism of hypoxia-induced up-regulation of FasL, we next examined whether hypoxia up-regulates FasL mRNA in EC. As shown in Figure 3, hypoxia induced a transient increase in the level of FasL mRNA at 3 hours.

Hypoxia up-regulates Fas ligand (FasL) expression in endothelial cells (EC). HUVEC were incubated for 6 hours under hypoxic conditions, and immunocytochemical studies were performed as described in “Materials and Methods.”

Hypoxia stimulates the expression of FasL in EC. A, membrane FasL (mFasL) expression by immunoblot analysis in HUVEC. HUVEC were exposed for various times (3, 6, 12, and 24 hours) to hypoxia (1% O2). Cell lysates (20 μg) were loaded onto 10% SDS-polyacrylamide gel and analyzed by immunoblotting using antihuman FasL monoclonal antibody. B, Effects of hypoxia on mFasL expression in EC with or without KB8301, a metalloproteinase inhibitor. HUVEC were exposed for 8 hours to hypoxia (1% O2). mFasL expression was analyzed with two different antibodies against FasL (N-20, NOK-1), as described in “Materials and Methods.” We used 1A12 cells (FasL-overexpressing cells) as a positive control.

Hypoxia up-regulates expression of FasL mRNA in EC. FasL mRNA expression in HUVEC was analyzed as described in “Materials and Methods.”
Hypoxia Up-Regulates sFasL Release
We next measured the levels of sFasL in the supernatant from EC. As shown in Figure 4A, the level of sFasL in the supernatant was increased after exposure to hypoxia at 12 hours. We observed the size of sFasL under nonreducing conditions, which shows an approximately 70-kd band, as an oligomeric complex (trimers) using an antibody against FasL, N-20 (Fig. 4A). To further quantify the level of sFasL, we also measured the level of sFasL in the supernatant by ELISA (Mochida Pharmacological, Tokyo, Japan). As shown in Figure 4B, hypoxia significantly increased the level of sFasL in the supernatant, which reached a peak at 12 hours and then returned to near the basal level at 24 hours.

Hypoxia stimulates sFasL release from EC. A, Changes in levels of sFasL released into the supernatant were determined by immunoblot analysis with an antibody against FasL (N-20). B, Time-course changes in concentration of sFasL in the supernatant from EC after exposure to hypoxia were determined by ELISA.
Hypoxia Down-Regulates FLIP Expression
To examine whether hypoxia can sensitize Fas-mediated EC apoptosis, we next measured the level of FADD-like interleukin-1β converting enzyme (FLICE)-inhibitory protein (FLIP), which is a recently identified as Fas-associated death domain-containing protein (FADD)-binding suppressor of apoptosis in EC. As shown in Figure 5, the expression of FLIP was significantly down-regulated from 3 hours after exposure to hypoxia in EC.

Hypoxia down-regulates expression of FLICE-inhibitory protein (FLIP) in EC. HUVEC were exposed for various times (3, 6, and 12 hours) to hypoxia (1% O2). Cell lysates (20 μg) were loaded onto 10% SDS-polyacrylamide gel and analyzed by immunoblotting using antihuman monoclonal antibody as described in “Materials and Methods.”
sFasL Released from EC Suppresses Hypoxia-Induced EC Apoptosis
We next examined whether sFasL released from EC modulates EC apoptosis induced by hypoxia. Incubation with the supernatant from EC exposed to hypoxia for 12 hours prevented EC from undergoing hypoxia-induced apoptosis (Fig. 6), whereas the supernatant from control EC had no effect (data not shown). However, the protective effect of the supernatant was partially inhibited by coincubation with a neutralizing antibody against FasL (Fig. 6).

Soluble Fas ligand (sFasL) released from EC inhibits hypoxia-induced EC apoptosis. HUVEC were incubated for 6 hours under hypoxic condition in the presence or absence of supernatants obtained from EC exposed for 12 hours to hypoxia. A neutralizing antibody against FasL (10 μg/ml) was added just before exposure to hypoxia. Apoptosis was then analyzed by a flow cytometer as described in “Materials and Methods.” Control versus hypoxia (* p < 0.05), hypoxia in the presence of supernatant versus in absence of the supernatant (** p < 0.01), hypoxia in the absence of anti-FasL IgG versus in the presence of anti-FasL IgG (+ p < 0.05).
An Inhibitor of Metalloproteinase Suppresses the Release of sFasL and Facilitates Hypoxia-Induced EC Apoptosis
KB8301, an inhibitor of metalloproteinase, partially suppressed the release of sFasL (Fig. 7A) and increased the level of cell-surface FasL (Fig. 7B). This KB8301-induced suppression of the sFasL release was confirmed by measuring sFasL concentrations in the supernatant by ELISA (data not shown). Although KB8301 itself did not induce apoptosis, it significantly enhanced hypoxia-induced EC apoptosis (Fig. 7C). In addition, exogenously added recombinant sFasL (10 ng/ml) significantly suppressed hypoxia-induced EC apoptosis in the presence or absence of KB8301.

Effects of sFasL and KB8301, a metalloproteinase inhibitor, on hypoxia-induced EC apoptosis. HUVEC were exposed for 12 hours to hypoxia (1% O2) in the presence or absence of KB8301 (10 μmol/L). Levels of sFasL released from EC (A) and mFasL (B) exposed to hypoxia were determined as described in “Materials and Methods.” C, EC apoptosis after exposure to hypoxia for 6 hours in the presence or absence of 10 μmol/L metalloproteinase inhibitor (KB8301) and with or without sFasL (10 ng/ml) was analyzed by flow cytometry as described in “Materials and Methods.” Control versus hypoxia (* p < 0.05), hypoxia in the absence of sFasL versus in the presence of sFasL (** p < 0.01), hypoxia in the absence of KB8301 versus in the presence of KB8301 (+ p < 0.05), hypoxia with KB8301 in the absence of sFasL versus in the presence of sFasL (++ p < 0.01). NS, not significant.
Discussion
In the present study, we demonstrated two major findings. First, we showed that hypoxia stimulated sFasL release by up-regulating FasL expression in EC. Second and most importantly, sFasL released from EC inhibited hypoxia-induced EC apoptosis. FasL, a member of the growing TNF family, is synthesized as a type II membrane protein that acts locally through cell-to-cell contact. sFasL is released from the cell surface as the result of metalloproteinase cleavage. sFasL has been detected in the serum of patients with myocarditis (Toyozaki et al, 1998) and congestive heart failure (Yamaguchi et al, 1999), in cerebrospinal fluid after severe head injury (Ertel et al, 1997), and in bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome (Matute Bello et al, 1999). In our study, the peak level of sFasL released from EC in the supernatant was estimated to be approximately 100 pg/ml by ELISA. Although the protective effect of recombinant sFasL at 10 ng/ml seems to be smaller than that of conditioned supernatant, this might be due to differences in oligomerization or binding affinity (Schneider et al, 1998). It is also possible that other factors, as well as sFasL, might be involved in the mechanism of the antiapoptotic effect induced by the supernatant.
FasL and TNFα are cleaved by distinct proteases with different substrate specificities. However, the cleavage of both FasL and TNFα is blocked by a matrix metalloproteinase inhibitor, suggesting that proteinases cleaving both cytokines carry a similar active site (Tanaka et al, 1998). Although both cell surface and secreted sFasL seem to be biologically active, the physiologic functions of sFasL have not been elucidated. Initially, it was speculated that sFasL might cause systemic tissue damage, because Fas is expressed in almost all tissues. However, the finding that sFasL is less cytotoxic than membrane-bound FasL (mFasL) suggests that conversion of mFasL to sFasL may prevent the killing of healthy bystander cells (Schneider et al, 1998). In the present study, we also showed that sFasL may act as an antiapoptotic molecule in FasL-expressing cells under hypoxic stress. Thus, the use of metalloproteinase inhibitors to treat tissue damage caused by hypoxia may result in an exacerbation of the disease through up-regulating mFasL expression. However, we cannot exclude that a high local concentration of sFasL may induce apoptosis in Fas-bearing cells.
EC are resistant to Fas-mediated apoptosis under normal conditions (Sata and Walsh, 1998a, 1998b). However, EC can be sensitized to Fas-mediated apoptosis under pathologic conditions (Suhara et al, 1998). In this study, we observed that hypoxia down-regulated the expression of FLIP that inhibits Fas-mediated apoptosis, indicating that Fas-mediated apoptosis may be sensitized in EC under hypoxic conditions.
Although the supernatant significantly suppressed hypoxia-induced EC apoptosis, hypoxia induced high levels of apoptosis in EC cultured without conditioned supernatants. This suggests that the concentration of sFasL released from EC may not reach a sufficient level to effectively protect against hypoxia-induced apoptosis. Although local tissue levels might be substantially higher than in tissue culture supernatants, more evidence is necessary to confirm whether hypoxia-induced sFasL shedding is really primarily an antiapoptotic mechanism in vivo.
There are at least two possibilities to explain the antiapoptotic action of sFasL. First, Fas could be quickly internalized with sFasL and down-regulated without inducing a sufficient level of caspase 8 activation (Tanaka et al, 1998). It is known that the ligation of Fas with FasL induces the formation of death-inducing signaling complex (DISC), which contains Fas, as well as several signaling components such as FADD and caspase 8 (Medema et al, 1997). It is believed that the DISC induced by mFasL may remain longer at the plasma membrane and result in a higher level of caspase 8 activation, whereas sFasL-induced caspase 8 activation may be minimal because Fas is quickly internalized with sFasL. Thus, down-regulation of Fas may be involved in the antiapoptotic action induced by sFasL. Second, sFasL/Fas complex could trigger signaling pathways that, rather than causing apoptosis, lead to activation of survival factors. In this regard, there is recent evidence that in certain cell lines, Fas-activation does not trigger apoptosis, but induces activation of nuclear factor-kappa B (NF-ϰB) and interleukin-8 secretion (Allison et al, 1997; Kang et al, 1997). Additionally, previous studies suggest that TNF activation of the NF-ϰB transcription factor family antagonizes the proapoptotic signals initiated by TNFα (Van Antwerp et al, 1996; Wang et al, 1998; Wu et al, 1998). Thus, activation of NF-ϰB could participate in the mechanism of the antiapoptotic function induced by sFasL. More recently, Shinohara et al (2000) also reported that Fas drives cell cycle progression in glioma cells via extracellular signal-regulated kinase activation. From these findings it may be interesting to examine whether sFasL can inhibit Fas-independent pathways. These experiments are now underway in our laboratory.
In conclusion, our results indicate that sFasL released from EC could be an adaptive feedback loop serving to prevent EC loss by apoptosis during hypoxic stress in the arterial wall.
Materials and Methods
Materials
Human recombinant sFasL and 1A12 cells, FasL-overexpressing mouse WR19L cells, were donated by Dr Shigekazu Nagata (Osaka University Medical School, Osaka, Japan). KB8301, a metalloproteinase inhibitor, was donated by Kanebo Inc. (Tokyo, Japan).
Cell Culture and Hypoxia Induction
HUVEC purchased from Sanko Junyaku (Tokyo, Japan) were cultured in CC-3156 EBM-2 (Sanko Junyaku) supplemented with 2% FBS and antibiotics without corticosteroid (Yang et al, 1995). For hypoxia experiments, confluent EC cultured in serum-free medium on 10-cm dishes were exposed for the indicated times to an ambient oxygen concentration of 1%, 5% CO2, and 94% N2 at 37° C (using a controlled incubator with CO2/O2 monitoring and CO2/N2 gas sources).
Immunocytochemical Assays
Cultured EC on a slide chamber (BIOCOAT; Becton Dickinson, Bedford, Massachusetts) were fixed in 4% paraformaldehyde for 20 minutes. After washing in PBS, endogenous peroxidase was blocked by immersing the samples in PBS containing 0.75% H2O2. To block nonspecific binding sites, the sections were incubated with blocking reagent (DAKO, Hamburg, Germany) for 10 minutes. The samples were then incubated overnight with the primary antibody against FasL (N-20; Santa Cruz Biotechnology, Santa Cruz, California) or negative IgG control. A mixture of antibodies against rabbit and mouse (DAKO) was added for 60 minutes. Finally, the samples were stained with the reagents of the DAB substrate kit (Dojindo Laboratories, Kumamoto, Japan). Positive labeling was stained brown-black.
Immunoblot Analysis
After exposure to hypoxia, cells were treated with 500 μl lysis buffer (50 mmol/L Tris-HCl [pH 8.0], 20 mmol/L EDTA, 1% SDS, 100 mmol/L NaCl). The supernatants (10 ml) from EC were concentrated to 100 μl with an Ultrafree-15, centrifugal filter device (Millipore, Bedford, Massachusetts). The cell lysates or supernatants (20 μg for each lane) were then analyzed by SDS PAGE using a 10% polyacrylamide gel and transferred to a PVDF membrane (Millipore). After blocking, the membrane was incubated with an antihuman FasL mouse monoclonal antibody (1:1,000, mAb33; Transduction Laboratories, Lexington, Kentucky), rabbit monoclonal antibody (1:500, N-20; Santa Cruz Biotechnology) or with an anti-FLIP rabbit antibody (donated by Kenneth Walsh, Tufts University School of Medicine, Boston, Massachusetts). Membranes were then washed and incubated with a horseradish peroxidase-conjugated mouse antibody (1:1,000, Promega, Madison, Wisconsin). After washing, FasL expression was detected using enhanced chemiluminescence (ECL; Amersham International, Buckinghamshire, United Kingdom). As loading controls we used detected loading proteins on the membranes stained with staining solution (7.5% acetic acid and 5% methanol).
RNA Isolation and RT-PCR
The expression of FasL mRNA was examined by RT-PCR analysis of total RNA from HUVEC and 1A12 cells (FasL-overexpressing T cells). Oligonucleotide primers were used in a quantitative RT-PCR assay: a sense primer (5′-GTTCTGGTTGCCTTGGTAGG-3′) and an antisense primer (5′-GACCAGAGAGAGCTCAGATACG-3′). As loading control primers, we used G3PDH oligonucleotide primers: a sense primer (5′-CTAGACCACAGTCCATGCCATCAC-3′) and an antisense primer (5′-CATGTAGGCCATGAGG- TCCACCAC-3′). Total RNA was extracted from EC using ISOGEN (Nippon Gene, Toyama, Japan). For RT-PCR, 1 μg total RNA was reverse-transcribed, incubated first at 42° C for 50 minutes and then at 99° C for 5 minutes. The reaction product was then subjected to PCR at 94° C for 2 minutes, followed by 32 cycles of 94° C for 40 seconds, 58° C for 60 seconds, and 72° C for 1.5 minutes. The FasL mRNA amplification products were separated by agarose gel electrophoresis and analyzed using ethidium bromide staining.
Flow Cytometric Analysis of FasL
After exposure to hypoxia, EC were washed twice with PBS containing 5% FCS. The cells were detached from dishes by addition of PBS containing 0.05% EDTA, without tripsinization. The cells (106) were stained first with 2 μg/ml human anti-FasL antibody (N-20 or NOK-1; PharMingen, San Diego, California) for 1 hour, and stained with a fluorescein isothiocyanate-conjugated antibody to rabbit or mouse IgG (Wako Junyaku, Osaka, Japan) for 30 minutes. FasL expression on the cell surface was then analyzed by FACSort (Becton Dickinson, Franklin Lakes, New Jersey).
Measurement of Concentration of sFasL
We measured the level of sFasL in the supernatant from EC by ELISA.
Apoptosis Detection Assay
Early apoptotic cells were detected with an antibody against annexin-V using MEBCYTO-Apoptosis Kit (Medical Biological Laboratories, Nagoya, Japan). Briefly, after hypoxic exposure, floating cells and adherent cells were collected and washed twice with cold PBS. Cells (106) were then incubated with a binding buffer and double-stained with propidium iodide and annexin V-fluorescein isothiocyanate-conjugated antibody. Propidium iodide and V-fluorescein isothiocyanate-conjugated antibody fluorescence on the cell surface were analyzed by FACSort (Becton Dickinson).
Statistical Analysis
Statistical analysis was performed by paired t test. Results are expressed as mean ± sem. A value of p < 0.05 was considered significant.
References
Allison J, Georgiou HM, Strasser A, and Vaux DL (1997). Transgenic expression of CD95 ligand on islet beta cells induces a granulocytic infiltration but does not confer immune privilege upon islet allografts. Proc Natl Acad Sci USA 94: 3943–3947.
Bombeli T, Karsan A, Tait JF, and Harlan JM (1997). Apoptotic vascular endothelial cells become procoagulant. Blood 89: 2429–2442.
Bombeli T, Schwartz BR, and Harlan JM (1999). Endothelial cells undergoing apoptosis become proadhesive for nonactivated platelets. Blood 93: 3831–3838.
Dimmeler S, Hermann C, and Zeiher AM (1998). Apoptosis of endothelial cells: Contribution to the pathophysiology of atherosclerosis? Eur Cytokine Netw 9: 697–698.
Ertel W, Keel M, Stocker R, Imhof HG, Leist M, Steckholzer U, Tanaka M, Trentz O, and Nagata S (1997). Detectable concentrations of Fas ligand in cerebrospinal fluid after severe head injury. J Neuroimmunol 80: 93–96.
Griffith TS, Brunner T, Fletcher SM, Green DR, and Ferguson TA (1995). Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 270: 1189–1192.
Hahne M, Rimoldi D, Schroter M, Romero P, Schreier M, French LE, Schneider P, Bornand T, Fontana A, Lienard D, Cerottini J, and Tschopp J (1996). Melanoma cell expression of Fas (Apo-1/CD95) ligand: Implications for tumor immune escape. Science 274: 1363–1366.
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, and Semenza GL (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev 12: 149–162.
Kang SM, Schneider DB, Lin Z, Hanahan D, Dichek DA, Stock PG, and Baekkeskov S (1997). Fas ligand expression in islets of Langerhans does not confer immune privilege and instead targets them for rapid destruction. Nat Med 3: 738–743.
Krammer PH, Dhein J, Walczak H, Behrmann I, Mariani S, Matiba B, Fath M, Daniel PT, Knipping E, and Westendorp MO (1994). The role of APO-1-mediated apoptosis in the immune system. Immunol Rev 142: 175–191.
Li D, Yang B, Philips MI, and Mehta JL (1999). Proapoptotic effects of ANG II in human coronary artery endothelial cells: Role of AT1 receptor and PKC activation. Am J Physiol 276: H786–H792.
Matute Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, and Martin TR (1999). Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 163: 2217–2225.
Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, and Peter ME (1997). FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J 16: 2794–2804.
Nagata S (1997). Apoptosis by death factor. Cell 88: 355–365.
Sata M and Walsh K (1998a). Oxidized LDL activates Fas-mediated endothelial cell apoptosis. J Clin Invest 102: 1682–1689.
Sata M and Walsh K (1998b). TNFα regulation of Fas ligand expression on the vascular endothelium modulates leukocyte extravasation. Nat Med 4: 415–420.
Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, Fontana A, and Tschopp J (1998). Conversion of membrane-bound Fas (CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med 187: 1205–1213.
Semenza GL, Roth PH, Fang HM, and Wang GL (1994). Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem 269: 23757–23763.
Shinohara H, Yagita H, Ikawa Y, and Oyaizu N (2000). Fas drives cell cycle progression in glioma cells via extracellular signal-regulated kinase activation. Cancer Res 60: 1766–1772.
Spyridopoulos I, Sullivan AB, Kearney M, Isner JM, and Losordo DW (1997). Estrogen-receptor-mediated inhibition of human endothelial cell apoptosis: Estradiol as a survival factor. Circulation 95: 1505–1514.
Stempien-Otero A, Karsan A, Cornejo CJ, Xiang H, Eunson T, Morrison RS, Kay M, Winn R, and Harlan J (1999). Mechanisms of hypoxia-induced endothelial cell death. Role of p53 in apoptosis. J Biol Chem 274: 8039–8045.
Strand S, Hofmann WJ, Hug H, Muller M, Otto G, Strand D, Mariani SM, Stremmel W, Krammer PH and Galle PR (1996). Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells. Nat Med 2: 1361–1366.
Suc I, Escargueil Blanc I, Troly M, Salvayre R, and Negre Salvayre A (1997). HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler Thromb Vasc Biol 17: 2158–2166.
Suhara T, Fukuo K, Sugimoto T, Morimoto S, Nakahashi T, Hata S, Shimizu M, and Ogihara T (1998). Hydrogen peroxide induces up-regulation of Fas in human endothelial cells. J Immunol 160: 4042–4047.
Tanaka M, Itai T, Adachi M, and Nagata S (1998). Downregulation of Fas ligand by shedding. Nat Med 4: 31–36.
Tanaka M, Suda T, Haze K, Nakamura N, Sato K, Kimura F, Motoyoshi K, Mizuki M, Tagawa S, Ohga S, Hatake K, Drummond AH, and Nagata S (1996). Fas ligand in human serum. Nat Med 2: 317–322.
Thompson CB (1995). Apoptosis in the pathogenesis and treatment of disease. Science 267: 1456–1462.
Toyozaki T, Hiroe M, Tanaka M, Nagata S, Ohwada H, and Marumo F (1998). Levels of soluble Fas ligand in myocarditis. Am J Cardiol 82: 246–248.
Van Antwerp DJ, Martin SJ, Kafri T, Green DR, and Verma IM (1996). Suppression of TNFα-induced apoptosis by NF-ϰB. Science 274: 787–789.
Wang CY, Mayo MW, Korneluk RG, Goeddel DV, and Baldwin AS Jr (1998). NF-ϰB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 281: 1680–1683.
Wenger RH and Gassmann M (1997). Oxygen(es) and the hypoxia-inducible factor-1. Biol Chem 378: 609–616.
Wu MX, Ao Z, Prasad KV, Wu R, and Schlossman SF (1998). IEX-1L, an apoptosis inhibitor involved in NF-ϰB-mediated cell survival. Science 281: 998–1001.
Yamaguchi S, Yamaoka M, Okuyama M, Nitoube J, Fukui A, Shirakabe M, Shirakawa K, Nakamura N, and Tomoike H (1999). Elevated circulating levels and cardiac secretion of soluble Fas ligand in patients with congestive heart failure. Am J Cardiol 83: 1500–1503, A8.
Yang Y, Mercep M, Ware CF, and Ashwell JD (1995). Fas and activation-induced Fas ligand mediate apoptosis of T cell hybridomas: Inhibition of Fas ligand expression by retinoic acid and glucocorticoids. J Exp Med 181: 1673–1682.
Acknowledgements
We thank Tomoko Hironaka for technical and secretarial assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mogi, M., Fukuo, K., Yang, J. et al. Hypoxia Stimulates Release of the Soluble Form of Fas Ligand That Inhibits Endothelial Cell Apoptosis. Lab Invest 81, 177–184 (2001). https://doi.org/10.1038/labinvest.3780225
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780225
This article is cited by
-
Expression of ADAM10, Fas, FasL and Soluble FasL in Patients with Oral Squamous Cell Carcinoma (OSCC) and their Association with Clinical-Pathological Parameters
Pathology & Oncology Research (2017)
-
Circulating levels of soluble Fas ligand reflect disease progression in multiple myeloma
Medical Oncology (2014)
-
Molecular ordering of hypoxia-induced apoptosis: critical involvement of the mitochondrial death pathway in a FADD/caspase-8 independent manner
Oncogene (2004)
