
Abstract
Genetic mechanisms leading to androgen-independent growth in advanced prostatic carcinomas (PC) are still poorly understood. Analysis of genes potentially involved in the regulation of tumor cell proliferation and apoptosis might confer better insight into this process and might lead to improved therapeutic strategies. Fluorescence in situ hybridization (FISH) analysis of dissociated nuclei with DNA probes for MYC (8q24)/#8, cyclin D1 gene (CCND1; 11q13)/#11, ERBB2 (17q13)/#17, the androgen receptor gene (AR; Xq12)/#X, and the retinoblastoma gene (RB; 13q14) was applied to formalin-fixed tissue from 63 patients with advanced PC after androgen deprivation therapy (ADT); matched tumor tissue before ADT was also available in 22 of these cases. The cut-points used were: “increased copy number,” ≥ 30% of all nuclei with increased FISH signals (centromere and/or gene); “amplification,” ≥ 15% of nuclei with “increased gene copy number.” CCND1 and MYC gene “amplifications” were present before ADT in 25% and 33% of the cases, respectively; the frequency of these “amplifications” increased to 37% and 57% after ADT. Loss of the RB gene was nearly four times more frequent after ADT than before therapy (22% versus 6%). AR and ERBB2 gene “amplifications” occurred only after ADT in 36% and 30% of cases, respectively. With the exception of the AR gene, the copy number increase was low. After treatment, MYC and AR gene “amplifications” correlated with the proliferation rate (Ki-67/MIB1 index; p = 0.01 and p = 0.04), whereas ERBB2 “amplifications” were associated with increased apoptotic index (PCD/TUNEL; p = 0.016). However, no correlation between FISH results and clinical follow-up could be established. FISH analysis of genes putatively involved in PC progression revealed characteristic patterns of aberrations in advanced PC before and after ADT. Distinct changes in gene copy number before and after therapy suggests possible involvement of these genes in the escape from androgen control.
Similar content being viewed by others


Introduction
Prostate cancer (PC) is one of the most common malignancies in the Western hemisphere (Mettlin, 1997). In Germany, PC is the third most common cause of male cancer mortality (Hölzel et al, 1996).
Androgens (testosterone and 5α-dihydrotestosterone) are essential for development, growth, differentiation, and maintenance of organ structure of the prostate. Physiologically, androgen deprivation leads to apoptosis (Isaacs et al, 1994). Initially, the growth of PC is androgen-dependent in the majority of cases. In locally advanced and/or metastatic PC, where prostatectomy is not curative, androgen deprivation therapy (ADT), performed either by castration or by other forms of endocrine manipulation (anti-androgens, luteinizing hormone-releasing hormone analogues), has been used for palliative therapy for nearly six decades (Huggins and Hodges, 1941). However, most cases become refractory to ADT within a few years.
As we could show recently by immunohistochemistry at the protein level, the physiologic regulation of proliferation and apoptosis is defective in the majority of advanced PC before ADT (Baretton et al, 1999). The genetic changes underlying androgen-independence and tumor progression are yet unclear. In the majority of previous publications, few, if any, gene amplifications could be found in PC (Latil et al, 1994; Fournier et al, 1995; Visakorpi et al, 1995a; Jenkins et al, 1997; Koivisto et al, 1997; Han et al, 1998). Moreover, the amplification rate of distinct oncogenes, such as ERBB2, is highly controversial (Kuhn et al, 1993; Ross et al, 1997a, 1997b). Very recently, Bubendorf and coworkers published a fluorescence in situ hybridization (FISH) study on consecutive formalin-fixed tissue microarray sections from a larger series of primary PC, as well as recurrent tumors and metastases from patients with hormone-refractory disease (Bubendorf et al, 1999). High-level amplifications were very rare in primary tumors (< 2%), but were higher in metastases and local recurrences (4% to 23%, depending on gene locus). However, because of inevitable nuclear truncations in 5-μm tissue sections, only high copy number gains were scored as specific amplifications in this FISH approach, and the number of tumor cell nuclei per microarray (diameter 0.6 mm) for statistical analysis is low.
To detect both low- and high-copy amplifications in a sufficient number of intact tumor cell nuclei, we analyzed aberrations of cMYC, cyclin-D1 (CCND1), HER-2/neu c-ERBB2 (ERBB2), the androgen receptor gene (AR), and the retinoblastoma gene (RB) by FISH on isolated nuclei from thick paraffin sections (20–25 μm). Furthermore, the correlation of the gene aberrations with tumor cell proliferation and apoptosis, as well as their prognostic impact, was tested before and after long-term ADT. The type of ADT applied was also included in the evaluation.
Results
FISH Analysis of Isolated Nuclei from Paraffin Blocks
Preparation of nuclear suspensions and their FISH analysis was successful for all DNA probes in both control cases with benign prostatic hyperplasia. As the FISH signal distributions showed, the hybridization efficiency of the probes was slightly different.
In the PC group, the FISH results were evaluable in 86% of cases for CCND1, in 91% of cases for MYC, in 88% of cases for RB, in 92% of cases for AR, and in 94% of cases for ERBB2. The hybridization efficiency of the probes was higher than 90%, but in some cases the amount of tumor tissue was insufficient for the investigation of all DNA probes.
FISH Analysis of PC before and after ADT
CCND1
Before ADT, 56% of cases showed a normal disomic signal pattern for CCND1 and centromere 11. In 19% of these cases, an increased copy number was detected. CCND1 gene “amplifications” were present in 25% of the tumors, including one PC with “high copy amplification.” After ADT, the frequency of “increased copy numbers” and “amplifications” of CCND1 increased to 24.5% and 37%, respectively (Figs. 1a and 2b). Table 1 shows the results of FISH analysis of paired specimens, cases after ADT only, and total number of cases after therapy.


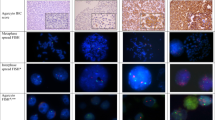
Fluorescence in situ hybridization (FISH) signal distributions for all cases that could be evaluated. Black, percentage of nuclei with gene “amplification” (ratio of gene copy number to centromere copy number ≥ 1.5). Dark gray, percentage of nuclei with “increased copy numbers” (ratio of gene copy number to centromere copy number = X/X); nuclei with increased centromere signals but decreased gene copy numbers were also included in this group (ratio of gene copy number to centromere copy number < 1, ie “relative gene copy deletion” relevant for ERBB2/#17 only). White, percentage of nuclei with normal, disomic FISH signals (ratio of gene copy number to centromere copy number = 2/2 or 1/1, respectively). Light gray, percentage of nuclei with centromere and/or gene copy number < 2 (not relevant for AR/#X). For the diagnosis “increased copy number,” at least 30% of nuclei had to show increased centromere and/or gene FISH signals. For the diagnosis “gene amplification,” at least half of these nuclei (15%) had to show a unbalanced increase of gene copy numbers (ratio of gene copy number to centromere copy number ≥ 1.5). a, Results for CCND1 and #11. b, Results for MYC and #8. c, Results for ERBB2 and #17. d, Results for AR and #X.

Images of FISH preparations on isolated nuclei from paraffin-embedded tumor material from patients with advanced prostatic carcinomas (Quips Genetic Workstation and Imaging Software, Vysis, Downers Grove, Illinois). a, MYC gene locus 8q24 (red) and centromere 8 (green). Left, a nucleus with normal, disomic FISH-signal distribution; right, a nucleus with MYC-gene amplification. b, CCND1 gene locus 11q13 (red) and centromere 11 (green). Upper left, a nucleus with normal, disomic FISH-signal distribution; upper right and lower left, tumor cell nuclei with balanced gains of CCND1-gene and centromere 11 signals; lower right, a nucleus with a high-copy CCND1 gene amplification. c, RB gene locus 13q14 (red). Above, a nucleus with normal, disomic FISH-signal distribution; below, three tumor cell nuclei with loss of one RB gene signal. d, ERBB2 gene locus 17q12 (red) and centromere 17 (green). Left, a nucleus with normal, disomic FISH-signal distribution (left nucleus) and a nucleus with “increased copy numbers” of one CCND1 and centromere 17 signal (right nucleus); right, a tumor cell nucleus with ERBB2 gene amplification. e, AR gene locus Xq12 (red) and centromere X (green). Upper left, a nucleus with normal, monosomic FISH signal distribution; lower left, a nucleus with “increased copy numbers” of one AR gene and centromere X signal; center and right, tumor cell nuclei with high-copy AR gene amplifications.
MYC Gene
Before ADT, 67% of the cases were normal for MYC and centromere 8 copy numbers. One-third of the tumors demonstrated an MYC gene “amplification” (all low-copy amplifications). After ADT, 11% of cases showed “increased copy numbers” of gene and centromere copy number, whereas 57% of PC exhibited MYC gene “amplifications,” including six cases (10%) with “high-copy amplifications” (Figs. 1b and 2a2a; Table 1).
RB Gene
Before ADT, a loss of the RB gene locus (Fig. 2c; Table 2) was observed in 6% of cases. Conversely, after ADT, 22% of cases showed loss of the RB gene locus. Gains of this chromosomal region were an occasional finding (in 6% of cases before ADT and in 15% after treatment).
ERBB2 Gene
Before ADT, 53% of tumors showed a normal disomic FISH signal distribution and 33% showed a balanced increase of gene and centromere copy numbers. No ERBB2 “amplifications” could be detected before ADT. In 14% of cases, a relative loss of the ERBB2 region was registered.
After ADT, 30% of tumors showed ERBB2 gene “amplifications,” including two cases with high gene copy numbers (3%). Forty-two percent of cases were disomic and in 13% a loss of ERBB2 was detected (Figs. 1c and 2d2d; Table 1).
AR Gene
Although a balanced increase of the AR gene locus and centromere X copies was seen before ADT (Table 1), AR gene amplifications occurred only after therapy (36% of cases, mostly high-copy amplifications) (Figs. 1d and 2e).
Correlation Between FISH Results and Proliferation (Ki-67/MIB1 Index) and Apoptosis (PCD Index)
No correlations between the results of the FISH analysis and proliferation or apoptosis could be found before ADT. After ADT, however, AR gene “amplifications” correlated with a higher maximal Ki-67 index, independent of the therapy regimen applied (41.7% ± 13.3 in cases with AR gene amplification versus 39.5% ± 26.0 in cases without AR gene “amplification”; p = 0.039; Mann-Whitney test).
Only tumors after conventional ADT showed a significant correlation between “amplifications” of the MYC locus and higher mean Ki-67 index (39.4% ± 19.7 in cases with MYC gene amplification versus 17.1% ± 19.0 in cases without MYC gene amplification; p = 0.01; Mann-Whitney test) and higher maximal Ki-67 index (51.1% ± 26.4 versus 22.5% ± 21.2; p = 0.012; Mann-Whitney test). ERBB2 gene “amplifications” were associated with increased apoptotic rate (programmed cell death [PCD] index) of the tumor cells (6.6% ± 5.4 in cases with ERBB2 gene “amplification” versus 2.8% ± 2.4 in cases without ERBB2 gene “amplification”; p = 0.016, Mann-Whitney test).
Correlation between FISH Results and Clinical Follow-Up
No correlations could be established between the results of the FISH analysis before or after ADT and the clinical course of the patients.
Discussion
The molecular mechanisms underlying development of androgen-independent growth in PC are still unclear. Our recent immunohistological data showed impaired regulation of tumor cell proliferation and apoptosis in most cases of advanced PC before and after ADT (Baretton et al, 1999). For a better understanding of the genetic changes leading to androgen-independent growth in PC, two oncogenes (cMYC, ERBB2), the cell cycle control gene CCND1, the androgen receptor (AR) gene, and the RB tumor-suppressor gene were studied by FISH analysis. Untreated primary tumors and recurrences after ADT were analyzed.
The selected dual labeling FISH technique (with the exception of the single labeling of the RB gene) on isolated tumor cell nuclei from previously microdissected paraffin-embedded tumor tissue allows the determination of subtle gene copy number aberrations on a cell-by-cell basis. The removal of any excess normal and reactive cells provides a high amount of intact tumor cell nuclei for FISH analysis and statistical evaluation. However, there are no defined cutoffs for FISH diagnosis of chromosomal and/or gene gains or losses in the literature. Thus, we are aware that our definition of gene aberrations, or “amplifications,” is more or less subjective. To make our data comparable with other studies, we show not only a final classification of the results (Table 1), but also present diagrams of the FISH-signal distributions per case (Fig. 1, a to d). As these diagrams show, a higher cut-point would have induced a lower frequency of gain for any given gene (Fig. 1, a to d), but the marked differences between before and after ADT would remain the same. For instance, no “amplifications” of the ERBB2 and the AR gene before ADT would result at any higher cut-point. On the other hand, lowering the cut-point to 10% would have added cases with “amplifications” before and after ADT, but again the differences between before and after treatment would not be significantly altered.
In accordance with a previous study (Baretton et al, 1994), the rate of increased or decreased chromosome and/or gene copies was very low in non-neoplastic prostatic tissue (Fig. 1, a to d) and the chosen cutoffs are far beyond these numbers. Therefore, we are confident that we have not overestimated the rate of chromosomal aberrations in our approach, for example, due to signal splitting of gene probes or the presence of G2-m-phase cells (Sauter et al, 1993, 1995).
Our results cannot be explained by an increase of the total number of chromosomal aberrations under ADT alone. The gene loci studied showed a rather characteristic pattern of gene copy number changes in PC before and after ADT.
MYC gene amplifications have been shown to play a role in solid tumors, for example, in colorectal and bladder carcinoma (Sauter et al, 1995; Donzelli et al, 1999). However, the importance of MYC for PC progression is still unclear. After long-term androgen deprivation, it could be shown that androgen receptor activity is increased and c-myc expression is altered in PC cells in vitro (Kokontis et al, 1994). The rate of 33% MYC gene “amplifications” before and 57% after ADT in our set of tumors agrees with other FISH studies. Two independent studies revealed a higher frequency of MYC gene amplifications in distant metastasis than in the primary tumor, suggesting an association between MYC and metastatic progression (Jenkins et al, 1997; Bubendorf et al, 1999). Moreover, substantial amplification of MYC was strongly correlated with increasing nuclear grade and immunohistochemical evidence of c-myc protein overexpression in one of these studies (Jenkins et al, 1997). We could show in our series that MYC gene “amplifications” correlate with proliferation rate of the tumor cells after conventional ADT. Data from studies using comparative genomic hybridization (CGH) indicate that MYC amplification is a potential marker of PC progression as well. With this technique, gains of 8q were revealed in 6% of PC cases before and 89% after ADT, respectively (Visakorpi et al, 1995b), and in 85% of metastatic PC (Cher et al, 1996).
The RB tumor suppressor gene located on 13q14 and CCND1 on 11q13 are both involved in the regulation of the cell cycle at the G1-S-phase transition. The rate of 6% losses of the RB gene FISH signals in PC before ADT and 22% after therapy in our study suggests an increasing impairment of cell cycle control. In CGH investigations, losses of the long arm of chromosome 13 were detected in 36% of cases before ADT and 56% after ADT (Visakorpi et al, 1995b), and in 75% of metastases (Cher et al, 1996).
CCND1 gene amplifications were described recently by Bubendorf and coworkers (Bubendorf et al, 1999) in 1.2% of primary tumors and in 4.7% to 7.9% of metastatic and recurrent PC, respectively. In our FISH study, “amplification” rates were higher before and after ADT (25% and 37%, respectively), which might be explained by the different materials (tissue sections in Bubendorf's study and isolated nuclei in our study). The majority of our aberrant cases exhibited only low copy CCND1 “amplifications.” These aberrations probably escape detection when thin sections are analyzed. The importance of CCND1 amplifications, however, is unclear, because no significant correlations to proliferation and apoptosis could be established. However, in vitro studies (Chen et al, 1998; Han et al, 1998; Perry et al, 1998) showed an influence of CCND1 on the development of androgen-independent growth. Since CCND1 levels are regulated post-translationally (Choi et al, 1997), gene aberrations might be less important for this effect.
The question of whether the ERBB2 gene is amplified in PC has been discussed in the literature. Whereas one group using solely a gene-specific FISH probe reports that ERBB2 amplification is a frequent genetic change in PC (44%) with prognostic impact (Ross et al, 1997a, 1997b), other groups did not detect ERBB2 amplifications at any stage of PC progression (Kuhn et al, 1993; Fournier et al, 1995; Bubendorf et al, 1999). Obviously, detection of ERBB2 amplifications in PC depends very much on technical factors (single or dual color FISH, sections or isolated nuclei). In our series, untreated PC showed either normal ERBB2 gene copy numbers or balanced increases of gene and centromere signals, whereas ERBB2 “amplifications” were present in 30% of cases after ADT. Only two cases, however, showed “high copy amplification.” Moreover, ERBB2 “amplifications” correlated with increased PCD index in cases after ADT. Thus, ERBB2 “amplifications” might be promoted by ADT.
In agreement with other FISH studies on hormone-refractory PC (Visakorpi et al, 1995a; Koivisto et al, 1997; Bubendorf et al, 1999), gains of the AR gene were the most common high-copy amplification. The frequency of 36% of AR gene “amplifications” also corresponds well with the results of these studies. In our set of tumors, AR “amplifications” could be found exclusively after ADT and correlated with a higher proliferation index of the tumor cells. This finding suggests a growth advantage for tumor cells with AR gene “amplification” under ADT. However, as for all other genes tested, no differences could be observed in the clinical follow-up of the patients.
In conclusion, the results of our FISH analysis of genes putatively involved in PC progression show characteristic patterns of aberrations before and after ADT. In particular, the occurrence of AR and ERBB2 gene “amplifications” after ADT suggest a possible involvement of these genes in escape from androgen control.
Materials and Methods
Materials and Patients
Formalin-fixed and paraffin-embedded tumor tissue from a total of 63 patients with locally advanced or metastasized primary PC was studied after ADT with and without additional therapy regimens (total of 85 samples). The tumors were graded histologically according to Gleason and Mellinger (Table 3) (Gleason, 1966; Gleason and Mellinger, 1974).
In 22 cases, paired specimens from both the untreated primary tumor and the recurrence after ADT were available (mean age of the patients at time of diagnosis, 72.3 years ± 8.3; mean duration of ADT, 40.9 months ± 34.7; median, 35.2 months; range, 0–121.5 months). In 12 of these cases, only a conventional ADT was performed either by orchiectomy (OE; n = 5), or by OE and drugs (n = 5), or only by drugs (n = 12). For the ADT by drugs, flutamide, fosfestrol, or cyproteronacetate were used primarily; later the patients were also treated with leuprorelinacetate, buserelinacetate, or goserelinacetate. In the remaining 10 cases, an additional chemotherapy (n = 5) or a non-specified form of therapy (n = 5) was applied. Estramustin, 4′-epirubicin, or cisplatin were used for chemotherapy.
In addition, tumor tissue from 41 patients after ADT was studied without paired material from the primary tumor. Twenty-three of these patients were pretreated only by conventional ADT (OE, n = 7; OE + drugs, n = 12; only drugs, n = 4). In the remaining 18 cases, an additional radiation (n = 4) or chemotherapy (n = 11), or a not specified therapy (n = 3) had been performed.
In 60 cases, clinical follow-up data were available, including all patients with paired tumor samples before and after ADT (mean time of observation, 62.8 ± 50.0 months; median, 50.6 months; range, 1–218 months). Twenty-eight of 60 patients died during the time of follow-up.
FISH
After removal of excess normal tissue under microscopic control, the tumor cell nuclei in the regions of interest were extracted from the paraffin blocks according to the method of Hyytinen et al (1994). Between the 20- to 25-μm-thick sections used for nuclear extraction 4-μm-thick hematoxylin-eosin-stained sections were prepared to ensure that representative tumor tissue was still preserved in the paraffin blocks.
Pretreatment
The cytospin slides were incubated in 4× SSC containing 0.1% Triton X for 1 hour (37° C). After washing in H2O, the slides were incubated in 1 m NaSCN (Sodiumisothiocyanate) for 30 minutes (56° C). After washing again in H2O, the slides were digested in Pepsin (1 mg/ml; Sigma, Deisenhofen, Germany) for 1 hour (37° C). The slides were then washed again in H2O and air-dried.
Denaturing steps
The slides were pre-denatured alone for 20 minutes at 80° C (heating plate). The hybridization mix was prepared according to the Vysis protocol: 10 μl of hybridization mix containing 7 μl hybridization buffer, 2 μl purified H2O, and 1 μl DNA probe, either MYC (8q24)/centromere 8, or CCND1 (11q13)/centromere 11, or ERBB2 (17q13)/centromere 17, or the androgen receptor gene (AR; Xq12)/centromere X, or the retinoblastoma gene/RB (13q14; all from Vysis, Downers Grove, Illinois); 2 μl of the hybridization mix was applied to the spots of nuclei. Slides were then denatured together with the hybridization mix at 80° C for 10 minutes, followed by overnight incubation under siliconized coverslips sealed with rubber cement in a humidified chamber at 37° C. The next day, the coverslips were removed by washing at room temperature in a buffer containing 0.5× SSC at 42° C for 10 minutes, followed by two additional washing steps in this buffer at 42° C for 10 minutes each. After air-drying, 4,5-diamino-2-phenylindol (DAPI, 1 mg/2.86 ml H2O)/Vectashield anti-fade solution (Vector Laboratories, Burlingame, California) was applied.
Evaluation of FISH Signals and Definitions of Gene Aberrations
The slides were evaluated under a Zeiss (Oberkochen, Germany) axioscope fluorescence microscope equipped with a HBO-100W mercury lamp and dual and triple band pass filters (Vysis). Hybridization signals were counted in 200 nuclei per case. Only discrete signals in non-overlapping nuclei with a distinct nuclear border were evaluated. Split signals were counted as one signal.
Chromosomal gains or losses were diagnosed if more than 30% of nuclei showed an increased or decreased number of FISH signals, respectively. Moreover, in the settings where both gene and centromere-specific probes were applied simultaneously for detection of oncogenes, we separated cases with “increased copy numbers,” that is, equal increases of gene and centromere signal number (ratio of gene copy number to centromere copy number = X/X), from cases with “gene amplifications,” that is, a disproportionate gain of gene copies. Thus, for the diagnosis of “gene amplification,” at least 30% of the nuclei had to show increased FISH signals and a ratio between gene and centromere copy numbers ≥ 1.5 had to be present in at least 15% of these nuclei. “High copy amplifications” exhibited innumerable gene signals (cutoff > 20 signals). To indicate that the terms “increased copy numbers,” “amplification,” and “high copy amplification” refer to our definitions, we use these terms in quotation marks throughout the text.
We are aware that these definitions are more or less subjective. The criteria for gene amplification, however, are not well established in the literature, nor is there a consensus as to what should be called “amplification.” Our main intention was to avoid an overestimation of gene amplifications and we are quite sure that our limits are high enough for this purpose. In this context, tissue samples of two transurethral resections from patients with benign prostatic hyperplasia served as controls, but were not used to establish cutoff values; the distributions of FISH signal counts in these cases (200 nuclei per probe) are also shown in Figure 1, a to d. As observed previously (Baretton et al, 1994), the rate of nuclei with increased gene copy number was consistently very low in benign prostatic hyperplasia (0% to 1.5%) and far below our limits for the diagnosis of gene aberrations. It cannot be excluded, however, that an underestimation of aberrations took place; therefore, we present not only a classification according to the limits mentioned above (Table 1), but also show the data as percentage FISH signal distribution per tumor for each gene (Fig. 1, a to d). This allows a comparison of our results with other studies in which different cutoff values are used.
Ki-67 Immunohistochemistry
Ki-67 immunohistochemistry was performed as previously described in detail (Baretton et al, 1999), using the MIB1/Ki-67 antibody (Dianova, Hamburg, Germany; dilution 1:1,000, prediluted in 1% bovine serum albumin; Sigma). According to the immunoreaction in 1,000 tumor cells per tumor, the mean and the maximal percentage of nuclei with a positive immunoreaction was determined (mean and maximal Ki-67 index).
Detection of Apoptotic Cells in Paraffin Sections
DNA fragments of apoptotic cells were visualized by an enzymatic reaction (Gavrieli et al, 1992; Ansari et al, 1993) as previously described in detail (Baretton et al, 1999). As internal positive controls, preserved non-neoplastic prostatic glands within the slides from primary tumors or lymphocytes in lymph follicles were used. Since the enzymatic reaction described labels both apoptotic cells and areas of necrosis, only those labeled cells were regarded as positive that showed additional characteristics of apoptosis, for example, isolated localization within an intact cell complex without an inflammatory reaction. At least 10,000 tumor cells per case were evaluated.
Statistical Analyses
Mean parameter values ± standard error of the mean (sem) were statistically compared by χ2 analysis, Mann-Whitney test, and the McNemar test (Hollander and Wolfe, 1973; Breslow and Day, 1980). Survival analyses were performed by Kaplan-Meier curves and the log rank test using the SPSS statistical software (SPSS Inc., Chicago, Illinois). p < 0.05 was regarded as statistically significant.
References
Ansari B, Coates PJ, Greenstein BD, and Hall PA (1993). In situ end-labeling detects DNA strand breaks in apoptosis and other physiological and pathological states. J Pathol 170: 1–8.
Baretton G, Valina C, Vogt T, Schneiderbanger K, Diebold J, and Löhrs U (1994). Interphase cytogenetic analysis of prostatic carcinomas by use of non-isotopic in situ hybridization. Cancer Res 54: 4472–4480.
Baretton GB, Klenk U, Diebold J, Schmeller N, and Löhrs U (1999). Proliferation- and apoptosis-associated factors in advanced prostatic carcinomas before and after androgen deprivation therapy: Prognostic significance of p21/WAF1/CIP1 expression. Br J Cancer 80: 546–555.
Breslow NE and Day NE (1980). The analysis of case control studies. IARC Scientific Publications, Lyon: No.32.
Bubendorf L, Kononen J, Koivisto P, Schraml P, Moch H, Gasser TC, Willi N, Mihatsch MJ, Sauter G, and Kallioniemi OP (1999). Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res 59: 803–806.
Chen Y, Martinez LA, LaCava M, Coghlan L, and Conti CJ (1998). Increased cell growth and tumorigenicity in human prostate LNCaP cells by overexpression to cyclin D1. Oncogene 16: 1913–1920.
Cher ML, Bova GS, Moore DH, Small EJ, Carroll PR, Pin SS, Epstein JI, Isaacs WB, and Jensen RH (1996). Genetic alterations in untreated metastases and androgen- independent prostate cancer detected by comparative genomic hybridization and allelotyping. Cancer Res 56: 3091–3102.
Choi YH, Lee SJ, Nguyen P, Jang JS, Lee J, Wu ML, Takano E, Maki M, Henkart PA, and Trepel JB (1997). Regulation of cyclin D1 by calpain protease. J Biol Chem 272: 28479–28484.
Donzelli M, Bernardi R, Negri C, Prosperi E, Padovan L, Lavialle C, Brison O, and Scovassi AI (1999). Apoptosis-prone phenotype of human colon carcinoma cells with a high amplification of the c-myc gene. Oncogene 18: 439–448.
Fournier G, Latil A, Amet Y, Abalain JH, Volant A, Mangin P, Floch HH, and Lidereau R (1995). Gene amplifications in advanced-stage human prostate cancer. Urol Res 22: 343–347.
Gavrieli Y, Sherman Y, and Ben-Sasson SA (1992). Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119: 493–501.
Gleason DF (1966). Classification of prostatic carcinomas. Cancer Chemother Rep 50: 125–128.
Gleason DF and Mellinger GI (1974). Veterans Administration Cooperative Urological Research Group. Prediction of prognosis for prostatic adenocarcinoma by combined histologic grading and clinical staging. J Urol 111: 58–64.
Han EK, Lim JT, Arber N, Rubin MA, Xing WQ, and Weinstein IB (1998). Cyclin D1 expression in human prostate carcinoma cell lines and primary tumors. Prostate 35: 95–101.
Hollander M and Wolfe DA (1973). Nonparametric statistical methods. New York: Wiley and Sons.
Hölzel D, Klamert A, and Schmidt M (1996). Krebs: Häufigkeiten, Befunde und Behandlungs-ergebnisse. Perspektiven für die Krebsdiskussion und eine quantitative klinisch-epidemiologische Onkologie aus dem Tumorregister München. München, Bern, Wien, New York: Zuckschwerdt.
Huggins C and Hodges CV (1941). Studies on prostatic cancer I. Cancer Res 1: 293–297.
Hyytinen E, Visakorpi T, Kallioniemi A, Kallioniemi OP, and Isola J (1994). Improved technique for analysis of formalin-fixed, paraffin-embedded tumors by fluorescence in situ hybridization. Cytometry 16: 93–99.
Isaacs JT, Furuya Y, and Berges R (1994). The role of androgen in the regulation of programmed cell death/apoptosis in normal and malignant prostatic tissue. Semin Cancer Biol 5: 391–400.
Jenkins RB, Qian J, Lieber MM, and Bostwick DG (1997). Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res 57: 524–531.
Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T, and Kallioniemi OP (1997). Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res 57: 314–319.
Kokontis J, Takakura K, Hay N, and Liao S (1994). Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res 54: 1566–1573.
Kuhn EJ, Kurnot RA, Sesterhenn IA, Chang EH, and Moul JW (1993). Expression of the c-erbB-2 (HER-2/neu) oncoprotein in human prostatic carcinoma. J Urol 150: 1427–1433.
Latil A, Baron JC, Cussenot O, Fournier G, Boccon GL, Le-Duc A, and Lidereau R (1994). Oncogene amplifications in early-stage human prostate carcinomas. Int. J Cancer 59: 637–638.
Mettlin C (1997). Clinical oncology update: Prostate cancer. Eur J Cancer 33: 340–347.
Perry JE, Grossmann ME, and Tindall DJ (1998). Epidermal growth factor induces cyclin D1 in a human prostate cancer cell line. Prostate 35: 117–124.
Ross JS, Sheehan C, Hayner BA, Ambros RA, Kallakury BV, Kaufman R, Fisher HA, and Muraca PJ (1997a). HER-2/neu gene amplification status in prostate cancer by fluorescence in situ hybridization. Hum Pathol 28: 827–833.
Ross JS, Sheehan CE, Hayner BA, Ambros RA, Kallakury BV, Kaufman-RP J, Fisher HA, Rifkin MD, and Muraca PJ (1997b). Prognostic significance of HER-2/neu gene amplification status by fluorescence in situ hybridization of prostate carcinoma. Cancer 79: 2162–2170.
Sauter G, Carroll P, Moch H, Kallioniemi A, Kerschmann R, Narayan P, Mihatsch MJ, and Waldman FM (1995). c-myc copy number gains in bladder cancer detected by fluorescence in situ hybridization. Am J Pathol 146: 1131–1139.
Sauter G, Moch H, Moore D, Carroll P, Kerschmann R, Chew K, Mihatsch MJ, Gudat F, and Waldman F (1993). Heterogeneity of erbB-2 gene amplification in bladder cancer. Cancer Res 53: 2199–2203.
Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, and Kallioniemi OP (1995a). In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 9: 401–406.
Visakorpi T, Kallioniemi AH, Syvänen A-C, Hyytinen ER, Karhu R, Tammela T, Isola J, and Kallioniemi OP (1995b). Genetic changes in primary and recurrent prostate cancer by comparative genomic hybridization. Cancer Res 55: 342–347.
Acknowledgements
The DNA probes were kindly provided by Vysis, Downers Grove, Illinois. The authors thank Nathalie Peterka for the excellent technical assistance in the laboratory work.
This paper was supported by a grant of the Deutsche Forschungsgemeinschaft (DFG-Nr. Ba 1458/2–1) to GBB.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper was presented in part at the United States and Canadian Academy of Pathology meeting in San Francisco, California, March 1999. The paper contains parts of AG's doctoral thesis.
Rights and permissions
About this article
Cite this article
Kaltz-Wittmer, C., Klenk, U., Glaessgen, A. et al. FISH Analysis of Gene Aberrations (MYC, CCND1, ERBB2, RB, and AR) in Advanced Prostatic Carcinomas Before and After Androgen Deprivation Therapy. Lab Invest 80, 1455–1464 (2000). https://doi.org/10.1038/labinvest.3780152
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780152
This article is cited by
-
A novel DNA methylation signature is associated with androgen receptor activity and patient prognosis in bone metastatic prostate cancer
Clinical Epigenetics (2021)
-
Pharmacological and genetic targeting of 5-lipoxygenase interrupts c-Myc oncogenic signaling and kills enzalutamide-resistant prostate cancer cells via apoptosis
Scientific Reports (2020)
-
Pathway-based expression profiling of benign prostatic hyperplasia and prostate cancer delineates an immunophilin molecule associated with cancer progression
Scientific Reports (2017)
-
Post genome-wide association studies functional characterization of prostate cancer risk loci
BMC Genomics (2013)
-
Tailoring to RB: tumour suppressor status and therapeutic response
Nature Reviews Cancer (2008)
