
Abstract
Experimental allergic encephalomyelitis (EAE) is a frequently employed animal model of the human disease multiple sclerosis. EAE can be induced by adoptive transfer of CD4+ T cells that are specific for central nervous system (CNS) antigens, typically myelin proteins. Although the pathogenic mechanism or mechanisms responsible for the clinical signs and histological changes in EAE and multiple sclerosis are not fully defined, the entry of T lymphocytes and antigen recognition within the CNS are required. The present study describes the participation of a novel cell surface molecule with properties suggesting a role in cell-cell adhesion or co-stimulation, or both, in the development of EAE in the rat. The molecule is defined by the unique monoclonal antibody (mAb) TLD-4A2. The TLD-4A2 antigen is present on resting and activated T lymphocytes, activated CNS endothelial cells, and microglia. The antigen is normally distributed in many tissues including lymph node, thymus, and spleen, as well as in the inflamed CNS. Both its pattern of tissue distribution and immunoprecipitation and immunoblotting studies suggest that the TLD-4A2 antigen is a novel molecule. Treatment of rats with the purified 4A2 mAb resulted in the inhibition of the clinical signs of EAE and also decreased the number T cells and macrophages accumulating in the CNS parenchyma. TLD-4A2 antibody did not seem to directly interfere with T cell viability in vivo, as demonstrated by the ability to recover and stimulate CD4+ encephalitogenic T cells from cervical lymph nodes of 4A2-treated animals. In vitro, the antibody partially blocked T cell proliferation assays. These data suggest that the TLD-4A2 mAb recognizes a novel molecule expressed on lymphocytes, endothelial cells, and macrophages that may play a role in hematogenous cell traffic and the initiation of CNS inflammation.
Similar content being viewed by others


Introduction
The central nervous system enjoys a relative state of immune privilege due in part to being sequestered from the immune system and being devoid of significant numbers of immune system cells (Barker and Billingham, 1977; Hickey et al, 1987, 1991; Wekerle et al, 1986). However, the brain is subject to a number of pathological inflammatory conditions wherein circulating leukocytes readily enter the central nervous system (CNS). Contrary to earlier notions concerning CNS immune privilege, there does exist a steady, albeit low, level of lymphocyte traffic through the normal brain and spinal cord (Hickey et al, 1987, 1991). In CNS inflammatory conditions—as also occurs in multiple sclerosis (MS), viral infections, and post-infectious inflammatory syndromes in humans, as well as animal model systems such as experimental allergic encephalomyelitis (EAE)—the level of T lymphocyte and macrophage entry into the CNS is greatly augmented (Hickey, 1991).
T lymphocytes and macrophages are the principal inflammatory cell types in MS and EAE lesions. The central role of these cells is demonstrated by studies employing monoclonal antibodies directed against a wide variety of molecules expressed on the surface of T lymphocytes and macrophages. Such treatment interferes with the function or migration of these cells, and blocks the initiation and development of EAE (Brosnan et al, 1981; Brostoff and Mason, 1984; Huitinga et al, 1990, 1993; Sriram and Steinman, 1983; Zaller et al, 1990). At this time, the exact parameters governing lymphocyte and macrophage traffic into the human CNS, as occurs in MS, are incompletely defined. Yet recent studies have underscored the importance of adhesion molecules and chemokine expression in such traffic (Baron et al, 1993; Hulkower et al, 1993; Kuchroo et al, 1993; Ransohoff et al, 1993; Yednock et al, 1992).
The analysis of EAE in rodent systems has helped delineate some of the requirements for T lymphocyte entry into the CNS needed to initiate inflammation. Of these, T cell activation is one of the most critical (Hickey et al, 1991; Kuchroo et al, 1993; Sloan et al, 1992; Wekerle et al, 1986; Yednock et al, 1992). Regardless of their antigen specificity, activated T cell blasts readily gain access to the CNS (Baron et al, 1993; Hickey, 1991; Hickey et al, 1991; Sloan et al, 1992; Wekerle et al, 1986). This observation supports the hypothesis that activation-related molecules on T cells and their counter-ligands on brain endothelium play a central role in T cell transendothelial migration (Archelos et al, 1993; Baron et al, 1993; Hickey 1991; Yednock et al, 1992). For T cells to produce inflammation in the CNS, the T lymphocyte must be specific for a CNS antigen and recognize that antigen in the context of major histocompatibility complex (MHC) class II molecules within the CNS (Hickey and Kimura, 1988; Hickey et al, 1991; Wekerle et al, 1986). The expression of accessory molecules by CNS glial cells may also contribute to T cell activation in the CNS and the subsequent appearance of inflammation (Williams et al, 1994). Despite these insights, our current state of understanding the identity, nature, and function of the specific cell surface molecules regulating T lymphocyte tissue entry and controlling effective antigen recognition is demonstrably incomplete.
For CNS inflammation, members of the monocyte/macrophage family are also critical. Two populations of brain macrophages are important in the pathogenesis of both MS and EAE: the perivascular macrophages and the parenchymal resident microglia (Graeber et al, 1992; Hickey, 1991; Hickey and Kimura, 1988; Williams et al, 1992). Both populations are bone marrow derived. The parenchymal microglia represent a stable population of CNS cells that have a very low level of turnover in rodents and humans (Hickey and Kimura, 1988; Lassmann et al, 1991; Unger et al, 1993). Perivascular macrophages are continuously repopulated from the bone marrow in rodents and humans (Hickey and Kimura, 1988). The perivascular macrophages have been shown to present antigen in vivo and are required for the initiation of CNS inflammation and induction of EAE (Hickey and Kimura, 1988). Both of these populations of CNS macrophages become activated in MS and EAE, and the attraction of additional circulating monocyte/macrophages to the CNS is significantly augmented during inflammation (Lassmann et al, 1991). Although the kinetics of macrophage turnover in the CNS have been studied, the specific parameters of macrophage traffic in normal and inflamed CNS, and the critical adhesion molecules and chemokines they require for participation in the lesions of MS and EAE, are not defined. Similar to T lymphocyte traffic, activation-induced molecules expressed by macrophages and their counter receptors on brain endothelial cells seem to be important in the migration process.
In this study, we characterize the expression of a novel activation-related cell surface molecule defined by the monoclonal antibody TLD-4A2, which was generated by immunizing mice with rat microglia (Flaris et al, 1993; Male et al, 1995). This antibody recognizes an antigen expressed on the majority of CD4+ T cells, certain subsets of monocytes, activated CNS endothelial cells, and reactive microglia (Male et al, 1995; Williams et al, 1996). The antibody partially inhibits antigen-stimulated T cell responses In vitro, and extensively inhibits the clinical manifestations of EAE as well as the accompanying CNS cellular infiltration. The studies described here support the contention that the TLD-4A2 molecule represents a heretofore undefined cell surface moiety involved in either cell migration or cell-cell signaling or both.
Results
Immunohistochemical characterization of 4A2 expression in normal tissues demonstrated diffuse staining in spleen, thymus, and lymph nodes. In confirmation of previous results (Male et al, 1995), the TLD-4A2 Ag was not detected in the spinal cord of normal control rats, but was demonstrated on CNS microglia and endothelial cells and inflammatory macrophages and T cells in spinal cord sections of rats with EAE (Fig. 1). This staining pattern and distribution on microglia, endothelium, macrophages, and T cells is unique for currently defined adhesion molecules.

4A2 Ag expression is upregulated in the central nervous system (CNS) in experimental allergic encephalomyelitis (EAE). Immunohistochemical staining of spinal cord tissue from rats injected with 5 × 106 encephalogenic peptide (EP)-specific CD4+ T cells. A, 4A2 antigen expression on inflammatory macrophages and T cells in the spinal cord of a rat with EAE. B, 4A2 antigen expression on CNS endothelium in the spinal cord of a rat with EAE. Original magnification, × 250.
Flow Cytometry
Flow cytometry studies demonstrated a high level of expression of the 4A2 antigen on both resting and activated encephalogenic peptide (EP)-specific T cells, and on many dissociated cells from the spleen, lymph node, and thymus of normal rats (Fig. 2). 4A2 antigen expression was readily detected on EP-specific T cell lines used in this study to transfer EAE. These cells were more than 94% TcR αβ+, and 95% CD4+ (data not shown). The expression of the 4A2 molecule was compared with the expression of CD4, MHC class I and II, VLA-4, and ICAM-1 by flow cytometry and noted to be distinct.

Flow cytometry comparisons of TLD-4A2, MHC class II, VLA-4, and ICAM-1 Ag expression on the EP T cell line. (The EP T cells were 94% TcR α, β+, and 95% CD4+ as assessed using flow cytometry.) The EP T cell line was stained under resting tissue culture conditions or after 3 days of ConA (5 μg/ml) activation in the presence of irradiated spleen accessory cells. Cells (1 × 106) were stained with a secondary isotype antibody control (20 Ab), TLD-4A2 mAb (7 μg/ml), OX-6 (MHC II) (50 μl of hybridoma supernate), anti-VLA-4 (10 μg/ml), and ICAM-1 (6 μg/ml) followed by goat anti-mouse IgG-FITC (1:650). Ten thousand events per stain were accumulated. Data demonstrate that the 4A2 molecule is expressed on more than 95% of EP T cells, lymph node cells, and cells from the thymus. The level of 4A2 expression is higher than that of MHC class II, VLA-4, and ICAM-1 on the same cells analyzed. Results presented here are representative of n = 2 flow cytometry experiments.
4A2 staining on resting EP-specific T cells was homotypic and virtually all cells were positive, to a greater extent than the staining for CD4 (not shown), TcR α, β (not shown), VLA-4, MHC class I and II, and ICAM-1 (Fig. 2). After mitogenic lectin (concanavalin-A) activation for 3 days, the expression of 4A2 antigen was decreased slightly on the same cells compared with the isotype control antibody and anti-T-cell receptor antibody that remained at approximately the same level. VLA-4, ICAM-1, and MHC class II antigen expression was upregulated on activated EP T cells. In confirmation of previous results (Male et al, 1995), 4A2 expression was demonstrated on the majority of cells from the lymph node (92%, n = 2 experiments). There was no demonstrated difference in 4A2 staining intensity between lymph node cells directly isolated versus lymph node cells that had been activated in culture for 3 days with ConA; however, ICAM-1, VLA-4, and MHC class II antigen expression was increased on lymph node cells after activation. In dissociated cells from the thymus, 4A2 antigen expression was the highest (92%) followed by VLA-4 (82%), MHC class II (61%), and ICAM-1 (42%). Approximately 50% of the cells in the spleen were 4A2 positive under basal conditions. This expression increased to more than 90% of the cells expressing the antigen after activation. A similar shift in positively stained cells was demonstrated with anti-ICAM-1, VLA-4, and MHC class II mAb.
Western Blot Studies
Figure 3 shows immunoprecipitates of the 4A2 Ag derived from brain endothelium and lymph node cells. The molecule appears as a major band at 50 kD with a minor band at 46 kD, both being single chain molecules. Molecular weights from endothelium and lymphocytes are similar. The precipitate derived from the endothelium also contained high-molecular-weight components (principally bovine IgG and C1q) from the plasma serum, in which the endothelial cells were grown. The components are seen in Figure 3, lane B at 220 kD. On the two-dimensional gel, these constituents are seen where IgG is 160 kD non-reduced, 55 kD and 25 kD reduced, and C1q is >220 kD non-reduced and has three closely set bands around 72 kD reduced.

Immunoprecipitation of 4A2 from an endothelial cell line and freshly derived lymph node cells. A, Immunoprecipitate from rat CNS endothelium (GP8. E4) in cell culture made with TLD-4A2, separated by two-dimensional SDS gel electrophoresis (non-reduced/reduced). The 4A2 protein appears as a major spot at 50 kD with a minor spot at 46 kD. The molecular weights are the same in both first (non-reduced) and second (reduced) dimensions. The major contaminant of the precipitate is IgG (160 kD non-reduced, 56 kD and 26 kD reduced). B, 4A2 immunoprecipitate (non-reduced) from brain endothelium. Major bands are at 50 kD and 46 kD, with high molecular weight contaminants. C, 4A2 immunoprecipitate (reduced) from rat lymph node cells with a major band at 50 kD.
Inhibition of EAE
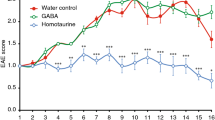
The injection of 5 × 106 CD4+ anti-EP T cells resulted in the induction of EAE that typically began by day 4 or 5 as evidenced clinically by a flaccid tail, and progressed to hind leg paralysis by day 5 or 6. In all studies, rats recovered by day 8 if not killed before that (Fig. 4). Clarified IgG1 ascites control rats receiving 1-mg doses of antibody 1 day before the injection of T cells, the day of T cell injection, and 2 days after T cell injection developed EAE similar to non-injected or saline-injected control rats. Treatment of rats with 1 mg of 4A2 1 day before T cell injection, the day of T cell injection, and 2 days post-injection (PI) resulted in either the total inhibition of EAE induction or a delay in the onset, and a reduction in the severity and duration of EAE (Fig. 4). The treatment of rats with a single dose (1 mg) of 4A2 2 days after injection of encephalitogenic cells also inhibited the onset and severity of EAE (n = 4 rats, two different experiments, two different EP T cell lines) (Fig. 4).

Treatment of rats with TLD-4A2 blocks or inhibits the induction and decreases the severity of EAE. Rats received 5 × 106 EP-specific T cells and were coded and assessed for the development of EAE. Rats were examined daily for the development of EAE, where a clinical score 0 = no clinical signs, 1 = flaccid tail, 2 = hind leg paralysis, and 3 = moribund state. Treatment groups consisted of rats receiving TLD-4A2 (4A2; —♦ —) (three doses at 1 mg per dose; 1 day before T cell injection, the day of T cell injection, and 2 days after T cell injection), rats receiving one injection of TLD-4A2 2 days after T cell injection (4A2 PI; —▪—), and control rats receiving either saline injections or IgG1 MOPC21 isotype control (EAE Control; —⊡—). The data presented here are the pooled results of n = 3 experiments, using n = 11 (controls), n = 12 (4A2-treated), and n = 4 (4A2 2 days PI) ± sem.
In three separate experiments using two different encephalitogenic, anti-EP, CD+4 T cell lines, 4A2 treatment resulted in either the complete block of EAE or a delayed onset and decreased severity of disease. In experiment 1, both the saline and ascites injected control animals developed flaccid tails and hind leg paralysis by day 6 PI (n = 5). In contrast, none of the 4A2-treated rats developed hind leg paralysis (n = 5) and only one of the 4A2 rats (20%, n = 5) developed tail flaccidity for 2 days, compared with the control rats (100%, n = 5) that had tail paralysis for 3 to 4 days. In experiments 2 and 3, using a different anti-EP T cell line, saline control rats had peak EAE at day 5 PI (n = 6 rats). On day 5 PI, 28% of the 4A2-treated rats developed tail flaccidity (n = 8) but none of these animals developed hind leg paralysis; 75% of the control animals had developed total hind leg paralysis (Table 1).
Taken together, all saline and MOPC control animals developed EAE, whereas only 50% of the 4A2-treated rats merely developed tail paralysis without hind leg paralysis. Eighty percent of the control rats reached the stage of hind limb paralysis (Table 1). There was a 4-fold increase in the maximal clinical severity in control rats as compared with the 4AZ–treated rats, and the duration of illness for 4A2 animals was 50% less than controls. Moreover, the average weight loss was 30% less for 4A2-treated animals than for controls (Table 1).
Flow cytometric studies demonstrate that the majority of anti-EP T cells were 4A2 immunoreactive. It is therefore possible that the antibody treatment inhibited the induction of EAE as a result of complement-mediated lysis of T cells, by opsonization of cells resulting in their removal from the circulation, or by inhibiting normal T cell function as a result of the induction of anergy or augmented activation. For this reason, control experiments to recover EP-specific cells were performed using 4A2-treated rats that did not develop EAE. In these experiments, the draining cervical lymph nodes (n = 5 rats) were removed on day 8 when animals were killed. Lymph nodes were dissociated and washed with Dulbecco’s phosphate–buffered saline (DPBS) and the cell suspension cultured in culture medium with 5% ConA medium and 20 μg/ml of EP peptide. Cells were cultured for 3 days and the resulting lymphoblasts were removed by Ficoll centrifugation. The recovered cells remained in culture for an additional 7 days until they returned to a resting morphology. Proliferation assays were performed in duplicate plates with triplicate wells per data point. The results demonstrate that viable EP-specific T cells were easily recovered from the cervical lymph nodes and that these cells could proliferate in response to their specific antigen (Fig. 5). The proliferative response was inhibited by preincubation of antigen presenting cells with the anti-MHC class II antibodies OX-3 and OX-6. Preincubation with 1 or 10 μg/ml of 4A2 mAb also resulted in partial inhibition of T cell proliferation to a greater extent than anti-VLA-4 (1 and 10 μg/ml) and similar to anti-ICAM-1 (1 and 10 μg/ml) treatment (Fig. 5). Pre-incubation with both TLD-4A2 (10 μg/ml) and ICAM-1 (10 μg/ml) almost completely inhibited T cell proliferation (82% inhibition, n = 3 experiments), similar to OX-3 and OX-6 controls.

Pre-incubation of TLD-4A2 inhibits proliferation of CD4+ T cells In vitro. Antigen presentation assays consisted of EP-specific CD4+ T cells (T) recovered from draining cervical lymph nodes of n = 5 rats that were treated with TLD-4A2 but did not develop EAE. EP-specific T cells were co-cultured with 2.5 × 105 irradiated lymph node and thymus cells. Cells were pulsed with EP peptide (20 μg/ml) and T cell proliferation assessed 48 hours later after a 16-hour pulse with 0.5 μCi 3H-thymidine. Inhibition studies were performed by pre-incubating TLD-4A2, anti-VLA-4, and anti-ICAM-1 (1 and 10 μg/ml) with irradiated lymph node and thymus cells for 1 hour before the addition of T cells. Positive controls for inhibition studies were OX-3 and OX-6 mAb (50 ml each) MHC class II blocking antibodies. The data are expressed as CPM of triplicate wells, and are representative of n = 4 experiments.
Histological Studies
Hematoxylin-eosin stained sections comparing spinal cord taken at the same time point from saline control rats with paralytic EAE and 4A2 treated animals that had not developed EAE showed perivascular inflammation. Lymphocytes and macrophages were evident in perivascular cuffs and in the CNS parenchyma of saline control rats that developed EAE. In the 4A2 mAb-treated animals, there were lower numbers of inflammatory cells and these were largely confined to perivascular cuffs with a few scattered cells in the parenchyma (Fig. 6). Immunohistochemical studies show a striking difference between cellular infiltrates using T cell and monocyte markers.

TLD-4A2 treatment inhibits the traffic of lymphocytes into the CNS parenchyma. Rats received 5 × 106 EP-specific T cells and were scored for the development of EAE. On day 5 the EAE saline-treated control rat (panel A) developed tail paralysis and was killed and compared with the TLD-4A2-treated rat that had no clinical signs (panel B). Hematoxylin-eosin staining demonstrates A, lymphocytic and macrophage infiltrate in perivascular and parenchymal locations on saline-treated control animals that developed EAE, and B, a lower number of infiltrates in the TLD-4A2 mAb-treated rats. Inflammation in these animals when found was always confined to perivascular locations. Results presented here are representative of n = 2 control and n = 5 experimental animals. Original magnification, × 250.
There was a marked difference in the amount of T cells in EAE versus TLD-4A2-treated animals using mAb R73 against αβ T cell receptor. As expected, T cells were undetectable in the normal CNS. In contrast, in the EAE animals, T cells were found throughout the spinal cord sections. 4A2-treated animals had a lower number of T cells than the control EAE animals, and the majority of these cells were restricted to the meningeal and sub-meningeal area.
Monoclonal antibodies against molecules expressed on macrophages and microglia also demonstrated a difference between the normal CNS, control animals that developed EAE, and 4A2-treated rats. It has been demonstrated that the mAb ED1 stains activated phagocytic macrophages and can be used to distinguish between infiltrating macrophages and phagocytic ameboid microglia in EAE versus resident resting microglia (Bauer et al, 1994). We found a striking difference between the number and distribution of ED1+ immunoreactive cells between the normal CNS, control EAE animals, and the 4A2-treated group. ED1 was not detectable in the normal CNS, whereas numerous ED1+ cells were demonstrated in perivascular cuffs and the parenchyma of the control EAE animals. The 4A2-treated animals had significantly lower numbers of ED1+ cells compared with the rats with EAE, and the majority of these cells were in the meninges with only a few scattered cells in the parenchyma (Fig. 7). OX-42, which is used as a marker of microglia and macrophages and also as an indicator of macrophage activation, further supported the notion that macrophages and microglia were highly activated in the EAE animals but not in the normal CNS or in 4A2-treated animals. In the normal CNS and the CNS of 4A2-treated animals, OX-42 staining was evenly distributed on microglia, revealing finely branched cells with crenulated processes. In contrast, OX-42 positivity in the CNS of EAE rats was accentuated on rounded, activated monocyte/macrophages in perivascular regions and in the parenchyma. The level of MHC class II expression, using the OX-3 and OX-6 mAb, also demonstrated a high level of induced MHC class II expression in the EAE animals over that of the non-inflamed CNS and 4A2-treated animals (Fig. 7).

Comparison of ED1, OX-42, and MHC class II antigen expression on rats that developed EAE versus their TLD-4A2-treated counterparts. The immunohistochemistry demonstrates A, ED1 reactivity of macrophage and microglia in EAE rat spinal cord compared with B, faint ED1 staining confined to meninges. C, OX-42 immune reactivity on activated rounded cells in the parenchyma compared with D, OX-42 staining of microglia in 4A2 treated spinal cord sections. This figure demonstrates resting microglia with branched processes. E, OX-6 immune reactivity on macrophage and T cells in the meninges and parenchyma of the spinal cord compared with F, OX-6 immune reactivity in the 4A2-treated rats that is found primarily in the meninges with scattered positive microglia in the parenchyma. Original magnification, all micrographs, × 250.
Blocking the cell surface molecule identified by the mAb TLD-4A2 resulted in inhibition of the clinical signs of EAE, a marked diminution of the parenchymal inflammation of the CNS, decreased CNS immune activation, and apparent failure of activated macrophages to migrate into spinal cord parenchyma. This was achieved without deleting or anergizing the encephalitogenic T cells that cause EAE.
Discussion
The data presented here describe an apparently novel monoclonal antibody defined molecule expressed on the majority of rat T cells that is also induced during inflammation on CNS endothelial cells, brain macrophages, and microglia. This molecule, defined by the TLD-4A2 mAb, appears to have a unique distribution. We have previously demonstrated that treatment of either T cells or endothelial cells with TLD-4A2 mAb increases binding of T cells to endothelium 4-fold (Male et al, 1995). This binding is inhibited with an anti-LFA-1 mAb or a combination of anti-LFA-1 and ICAM-1. Thus it appears that the TLD-4A2 antigen functions as a signaling molecule that augments integrin-mediated binding of lymphocytes to endothelium (Male et al, 1995).
In the current study, we found that pretreatment of rats with TLD-4A2 before the injection of encephalitogenic T cells, or 2 days after T cell infusion, results in either the complete inhibition of EAE or a significant delay in the onset of disease and a decrease in its severity. Results of antigen presentation studies support a role for the 4A2 molecule on CD4+ T cells and APC in T cell activation.
The data comparing the pattern and distribution of TLD-4A2 staining in a variety of tissues, as well as Western blot and flow cytometric studies, all support the notion that the 50 kDa single chain molecule recognized by TLD-4A2 is unique and not currently defined in the rat. Comparisons of 4A2 antigen with other defined molecules demonstrate that the 4A2 molecule is not similar to any of the other cell surface ligands previously demonstrated as involved in the induction of EAE. Moreover, comparison of the molecular weight and cellular distribution of the 4A2 antigen with what is known concerning the defined “CD” molecules fails to identify a similar entity in rodents and humans (Barclay et al, 1994; Pigott and Power, 1994).
Immunohistochemical studies of the CNS from control rats with EAE consistently demonstrated lymphocyte infiltration of the meninges, perivascular area, and extension into the neural parenchyma. In contrast, 4A2 mAb-treated rats that did not demonstrate clinical EAE had lower numbers of lymphocytes within the meninges, and diminished lymphocyte and macrophage accumulation within perivascular cuffs. Most notably, there were very few inflammatory foci within the parenchyma of the treated animals.
The role of infiltrating CD4+ T cells in producing the pathological changes seen in CNS inflammatory events is not fully defined (Williams et al, 1994b). Although myelin-reactive CD4+ T cells are not thought to be directly cytotoxic in vivo, they contribute to CNS inflammation indirectly by inducing CNS endothelial and glial cell activation, and by stimulating the recruitment of additional inflammatory cells to the nervous system. A number of earlier studies have employed mAb to interfere with T cell/endothelial cell or T cell/APC interactions and thereby block EAE induction (Baron et al, 1993; Brostoff and Mason, 1984; Kent et al, 1995; Sloan et al, 1992; Sriram and Roberts, 1986; Sriram and Steinman, 1983; Yednock et al, 1992; Zaller et al, 1990). With notable exceptions, few studies have investigated the mechanism(s) by which this EAE inhibition occurred (Bauer et al, 1995; Kent et al, 1995; Yednock et al, 1992).
Although we have not totally delineated the means by which TLD-4A2 mAb treatment interferes with the pathogenesis of EAE, the preponderance of evidence suggests that the 4A2 treatment results in the inability of T cells and macrophages to enter into the CNS parenchyma. It does not delete or anergize the encephalitogenic T cells, nor does it completely inhibit the ability of the T cells to respond to their specific antigen. It is known that T cell traffic to the CNS is required for the induction of EAE and most likely also for macrophage transmigration. The antibody treatment might interfere with disease initiation by blocking T cell/brain endothelial cell interaction and transmigration, or by inhibiting T cell and macrophage co-stimulation at the blood-brain barrier, thereby inhibiting secondary activation of one, the other, or both cell types.
What is the significance of 4A2 expression by immune system cells and CNS glial cells? Similar to LFA-1, ICAM-1, and MHC class II, the wide distribution of 4A2 within the immune organs, on microglia, and on the majority of rat T cells In vitro suggests an important involvement in intercellular interactions or cell migration or both.
The TLD-4A2 mAb was derived by immunizing mice with cultured rat microglia. Although we do not detect the 4A2 antigen in the normal, non-inflamed CNS, it is readily detected in the CNS of EAE animals as was shown in this study. Interestingly, the expression of 4A2 antigen in the CNS is seen within 48 hours after injection of encephalitogenic T cells; this is still 2 days before the animal becomes clinically ill and before histologically identifiable inflammation is noted (Williams et al, 1996). The de novo expression of the 4A2 antigen in the CNS appears to be activation-dependent. Whether the appearance of 4A2 in the CNS after EAE induction represents the actual de novo expression of the antigen or results from the unmasking of an activation dependent epitope or a constitutively expressed molecule is unknown.
Results presented in this study, and in previous publications, support the notion that the induction of 4A2 expression within the CNS is dependent upon the presence of T cells, possibly of the TH1 phenotype (Male et al, 1995; Williams et al, 1996). Microglial 4A2 expression is not detected in CNS stab wound injury or Wallerian degeneration after optic nerve transection, which in contrast to the EAE model are not immunologically mediated (Flaris et al, 1993; Molleston et al, 1993). Although there is glial cell and presumably brain endothelial cell activation in both the stab wound and Wallerian degeneration models, there are few inflammatory cells entering into the CNS from the peripheral immune system (Male et al, 1995; Williams et al, 1996). This observation, coupled with the evidence that 4A2 expression is increased on brain endothelial cells after IFN-γ and not after TNF incubation, supports the contention that T cells of the TH1 phenotype may be responsible for the induction of the 4A2 Ag in EAE (Male et al, 1995). Thus the 4A2 molecule may be a necessary moiety for T lymphocyte and macrophage traffic into the CNS. It also is a marker of brain endothelial and macrophage and microglial cell activation in delayed type hypersensitivity (DTH) responses mediated by CD4+ T cells of the TH1 phenotype.
There are several putative mechanisms by which the TLD-4A2 mAb might inhibit the induction of EAE in the adoptive transfer model. We have addressed most of these in control studies. Hematoxylin-eosin stained sections from rats treated with the 4A2 antibody, but not given encephalitogenic T cells, did not demonstrate apparent tissue damage due to the antibody treatment, nor was there evidence of endothelial cell damage or cellular depletion in lymphoid organs. Examination of 4A2-treated rats versus saline-injected controls revealed the expected lymphocyte and macrophage populations in both groups, however inflammatory cells were not found in the CNS parenchyma of 4A2 antibody-treated rats. This evidence and the demonstrated ability to recover EP-antigen-specific T cells from the cervical lymph nodes of 4A2-treated rats argue against the possibility that antibody treatment merely resulted in the opsonization and removal of the pathogenic T cells from the circulation. The antibody does not totally block antigen-specific T cell proliferation as seen in proliferation assays, however antibody treatment in vivo may sufficiently interfere with T cell and macrophage activation so that overt disease is abrogated.
It does not appear that TLD-4A2 mAb inhibits EAE development solely by blocking the entrance of encephalitogenic T cells into the target organ. Support for this is given by the experimental modification in which 4A2 was administered 2 days after the injection of encephalitogenic cells. It has been shown that CNS-specific T cells have already entered the CNS within 12 to 24 hours after intravenous infusion (Hickey et al, 1991). It is interesting to note that in our study, similar to Yednock et al, (1992), treatment with mAb at 48 hours after cellular injection also significantly inhibits or delays EAE development, severity, and length of illness. These data support the notion that TLD-4A2, at least in part, inhibits the non-specific recruitment or transendothelial migration of additional leukocytes needed to produce disease.
Because TLD-4A2 partially inhibits antigen-specific T cell stimulation In vitro, we cannot rule out that blockade of the 4A2 molecule interferes with a secondary (co-stimulation) signal that encephalitogenic T cells may require for initiating the tissue-specific inflammatory cascade. In the T cell stimulation assays employed here, irradiated thymocytes and lymph node cells were used as APC; antigen presentation assays using CNS-derived monocyte/macrophage because APC have not been performed. Therefore, it remains possible that the 4A2 antigen on such specialized, tissue-resident cells subserves a different or relatively more important function in the pathogenesis of inflammation. Likewise, it is possible that TLD-4A2 mAb could inhibit T cell-mediated macrophage stimulation. Overall, the bulk of the evidence suggests that treatment with the 4A2 mAb interferes with recruitment of lymphocytes or macrophages or both into the CNS and thereby impedes development of inflammation.
In addition to the decreased incidence of inflammatory cells, and possibly as a result of it, 4A2 treatment apparently impedes monocyte/macrophage and glial cell activation, as detected by significantly diminished levels MHC class II, OX-42, CD4, and ED1 molecule expression in the CNS of treated rats. ED1 stains the phagosomes of activated, phagocytic macrophages (Bauer et al, 1994; Damoiseaux et al, 1994). It has been shown that in EAE the majority of cells that are ED1+ are recently infiltrating macrophages with a smaller population of microglia that have become activated (Bauer et al, 1994). In addition, OX-42 is frequently used as an activation marker on microglia and macrophages (Graeber et al, 1990). The expression of CR3 (OX-42) is enhanced in rodent injury models and during the course of EAE (Molleston et al, 1993). After 4A2 treatment, there are markedly diminished levels of ED1 and OX-42 staining intensity compared with untreated animals with EAE, suggesting that it permitted a very low level of macrophage infiltration and parenchymal glial activation. The expression of MHC II molecules also define a distinct difference in immune activation of the parenchyma between the two treatment groups. In the 4A2 mAb-treated animals, there was little MHC class II expression compared with controls. The lower MHC class II expression by parenchymal microglial cells in the 4A2 treated animals is probably the result of the inhibition of T cell traffic into the parenchyma and thus a diminished amount of IFN-γ to which they are exposed.
Although we do not have direct evidence at this time of what exactly the 4A2 molecule is, we do know from this study what molecules it is not. Based on immunohistochemical and immune precipitation data, it is clear that the 4A2 molecule has not previously been demonstrated. Several lines of evidence support our contention. Similar to this study using TLD-4A2 mAb, several earlier reports have demonstrated EAE inhibition using purified mAb against a series of immunologically important molecules including: MHC class II molecules (Sriram and Steinman, 1983; Steinman et al, 1981), the T cell receptor (Zaller et al, 1990), CD4, ICAM-1 (Archelos et al, 1993), VLA-4 (Baron et al, 1993; Kent et al, 1995; Yednock et al, 1992) CR3 (Huitinga et al, 1993), CD28 (Cross et al, 1990), and the CD40L (gp39) (Gerrotse et al, 1996). Yet all of these other molecules have molecular weights, cellular distributions, and flow cytometry profiles distinct from the 4A2 molecule. CR3, CD28, and CD4 are not expressed by CNS endothelial cells. Flow cytometric results of freshly derived or activated cells from the spleen, thymus, and lymph node, and resting or activated CD4+ T cell lines demonstrate that the 4A2 mAb has a staining profile different from MHC class II mAb, CD4, VLA-4, and ICAM-1 profiles. Although CD40 is induced on endothelial cells and expressed on perivascular macrophages in MS and EAE, it is not demonstrated on T cell lines In vitro. The CD40 molecule is detected on T cell lines after antigen or mitogenic stimulation (Gerrotse et al 1996).
Candidate molecules that have a similar distribution but have not been implicated in contributing to EAE induction include CD11c (LFA-1), CD31 (PECAM), and CD44 (gp-90, HERMES). The results of immunoblotting studies indicate that the TLD-4A2 mAb recognizes a single chain molecule that is 50 kD under reducing and non-reducing conditions. These sizes are different from the molecular weight (MW) reported for CD11a (150, 170, and 180 kD reduced; 145, 165, 170 kD non-reduced; Larson and Springer, 1990), CD31 (130 to 140 kD reduced and non-reduced), and CD44 (80 to 95 kD and 130 kD reduced; Stockinger et al, 1990). However, the MW reported for different molecules may vary considerably depending on tissue source of the protein and also the species from which the protein was derived (Shimizu and Shaw, 1991). The known distribution of LFA-1, PECAM-1, and CD44 in tissues also argues against these molecules being identified by TLD-4A2. CD11a is primarily a leukocyte antigen and is not expressed to a significant level on rat CNS endothelial cells (Cannella et al, 1990). CD31 was a potential candidate molecule for 4A2, and PECAM-1 is expressed on rat brain endothelial cells and T cells (Jackson et al, 1992). But the expression of CD31 on microglia has not been demonstrated. Furthermore, we have screened the TLD-4A2 mAb against recombinant rat PECAM-1 and did not find reactivity by ELISA—although other monoclonal antibodies of the TLD series were CD31-specific (Male et al, 1995). Similar to the expression of PECAM-1, CD44 is expressed on astrocytes in the brain but is not reportedly expressed on microglia (Aho et al, 1994; Moretto et al, 1993). Thus, the bulk of currently available evidence suggests that this is a novel cell surface molecule (Barclay et al, 1994; Pigott and Power, 1994).
The data presented in this study demonstrate that the TLD-4A2-defined molecule is a novel, functional cell surface moiety expressed by cells of the immune system and activated endothelial cells and microglia. Functionally, this molecule plays a role in T lymphocytes binding to endothelial cells In vitro. Moreover, antibodies against the molecule, when administered to rats due to develop EAE, prevent the disease presumably by inhibiting lymphocyte and macrophage migration into the CNS. The 4A2 antigen is an important new molecule with adhesion or co-stimulatory properties that are critical to the development of chronic inflammatory conditions of a type such as EAE. It is probable that the homolog of this cell surface molecule exists in humans. It might serve as an important immune-regulatory molecule in human illnesses, and may be an appropriate target for therapeutic manipulation.
Materials and Methods
Experimental Animals
Female Lewis rats, 2 to 3 months of age (n = 27), were obtained from Charles River Laboratories (Wilmington, Massachusetts) and housed in the animal facilities at Dartmouth Medical School, and the New England Regional Primate Research Center, Harvard Medical School. Animals were cared for in accordance with National Institutes of Health guidelines on laboratory animal welfare.
Immunization and Cell Line Preparation
For these studies, cell lines were established that were specific for an encephalitogenic peptide (EP) of myelin-basic protein (Mannie et al, 1985). The EP was dissolved in PBS and emulsified with an equal volume of complete Freund’a adjuvant (CFA) supplemented with 5.0 mg/ml Mycobacterium tuberculosis H37RA (DIFCO, Detroit, Michigan) for a final concentration of 1 mg/ml of EP. The EP-CFA emulsion was injected intradermally in the footpad of Lewis rats (200 ug per animal). Nine days after immunization, draining popliteal and inguinal lymph nodes were isolated, dissociated under sterile conditions, and the lymphocytes were cultured in the presence of EP at 20 μg/ml final volume using initiation medium. Initiation medium consists of RPMI 1640 supplemented with 1% heat inactivated rat serum, 5% NCTC-109, 5 × 10−5M 2-mercaptoethanol, 2 mm glutamine, 100 μg/ml penicillin, 100 ug/ul streptomycin, and 100 μg/ml fungizone. After 3 days of antigen stimulation, antigen-specific blasts were separated using Histopaque 1.077 (Sigma Chemical Co., St. Louis, Missouri). T lymphocytes were recovered from the density interface, washed twice with PBS, and expanded in a culture medium similar to that previously described, but with 10% FBS (HyCLone Laboratories, Logan, Utah) instead of 1% rat serum. Antigen specificity assays were performed as described below.
TLD-4A2 Preparation and Purification
The TLD-4A2 mAb was derived after immunization of mice with rat microglia cells (Flaris et al, 1993). The isotype of the TLD-4A2 mAb is IgG1 as was determined by immune precipitation in gels using a kit (Male et al, 1995). Ascites were produced in pristane-treated Balb/cJ mice and the gamma globulin fractions where purified by precipitation with a saturated ammonium sulfate solution. The gamma globulin fraction was dialyzed against PBS for 48 hours with several PBS changes, and then sterile filtered with a 0.45 mm low protein binding filter (Corning, Corning, New York). The concentration of the mAb was adjusted to 1 μg/ml of protein with PBS.
Induction of EAE and Antibody Treatment
EAE was induced by tail vein injection of EP-specific T lymphoblasts (5 × 106 cells per animal) that had been activated In vitro for 3 days with the mitogenic lectin Concanavalin A (5 μg/ml) (Sigma) in the presence of irradiated (1,500 rad) spleen accessory cells (Hickey et al, 1987). Experimental animals received EP-specific, encephalitogenic T cells, and either: (a) 1 ml saline; (b) 1 mg clarified ascites MOPC-21 (IgG1 isotype control, diluted in PBS for final concentration 1 mg/ml) (Sigma); or (c) 1 mg mAb TLD-4A2 purified from ascites and diluted in PBS to a final concentration 1 mg/ml. Rats received injections of saline or antibody ip 1 day before the injection of encephalitogenic T cells, the day of T cell infusion, and 2 days after T cell injection. In two additional experiments, rats (n = 4) received a single dose of 4A2 antibody 2 days after the injection of EP-specific T cells. Rats in the different treatment groups were coded, weighed, and assessed for clinical scores in a blind fashion. Clinical scores are as follows: 0, no disease; 1, flaccid tail; 2, hind limb paralysis; and 3, paralysis and exhibiting a moribund state (Hickey et al, 1983, 1987).
Histological Studies
For selected rats used in the study, brain, spinal cord, spleen, thymus, and lymph nodes were removed, either at peak incidence of disease (day 4 to 6) or after recovery, and fixed in 10% formalin. Paraffin-embedded tissues were cut and stained with hematoxylin-eosin for histological examination. Control rats, given saline or control immune globulin, were killed and compared with 4A2-treated rats that had no clinical signs. Two rats that received three, 1 mg/ml injections of the TLD-4A2 mAb, but not encephalitogenic T cells, were killed 7 days after the initial antibody injection and their thymus, spleen, lymph nodes, and CNS tissues were prepared for histological study. Tissues were processed for immunohistochemistry as described below.
Antibodies and Immunocytochemistry
For these studies, control rats with EAE and TLD-4A2 mAb-treated rats that did not develop disease were deeply anesthetized and perfused via the aorta with calcium/magnesium free PBS, followed by 200 ml of 2% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Tissues were removed and post-fixed in 2% paraformaldehyde for 1 hour. Thereafter, tissues underwent extensive washes with PBS, and were cryoprotected in a 30% sucrose/PBS (v/v) solution overnight at 4° C. The next day, tissues were blotted dry, blocked, and frozen in O.C.T. Compound (Miles Inc., Elkhart, IN). Cryostat sections 6-μm thick were cut and slides allowed to air dry. The staining procedure used has been described in detail elsewhere (Williams et al, 1996). Briefly, sections were rinsed with 0.5 M Tris buffer, pH 7.6, and then sections were incubated with appropriate primary mAb overnight at 4° C. The next day, slides were incubated with a biotinylated secondary anti-mouse IgG that had been pre-absorbed against rat tissues (Vector Laboratories, Burlingame, California) overnight. Sections were then washed three times with PBS and incubated with avidin-biotinylated horseradish peroxidase complex (ABC; Vector Laboratories) for 2 hours. The color reaction product was developed using 3,3′-diaminobenzidine in the presence of H2O2. Sections were dehydrated through a series of ethanols and xylene, coverslipped, and assessed histologically.
A series of cell lineage and MHC specific antibodies were used for immunohistochemical studies on tissue sections and for flow cytometry analysis. The antibodies and their specificity are OX-42 (CD 11 b/c, IgG 2a), OX3 and OX-6 (anti-MHC class II I-A, IgG1), W3/25 (anti-rat CD4, IgG1), OX-8 (anti-rat CD8, IgG1), R73 (anti rat TCR-alpha IgG1) purchased from Serotec (Oxford, United Kingdom). An anti-alpha 4 integrin (VLA-4; a gift from Ted Yednock, Athena Neurosciences, South San Francisco, California; clone L-25, IgG2b) anti-ICAM-1 (a gift from Ted Yednock; clone TM8, IgG1), and TLD-4A2 were also used for flow cytometry and for studies on the inhibition of antigen-specific T cell proliferation.
Flow Cytometry
Flow cytometry studies were undertaken to assess the relative expression of the 4A2 molecule on resting and activated anti-EP T cells compared with a variety of T cell surface molecules, including CD4, MHC class II, VLA-4, and ICAM-1. The same antibodies were used to define antigen expression on dissociated spleen cells, and cells from the lymph node and thymus. Cells were stained either immediately after tissue dissociation or after 3 days of In vitro activation. Dissociated cells and cell lines were counted and adjusted to 0.5 to 1 × 106 cells per milliliter per data point. Controls consisted of non-stained cells, exposed to MOPC21 (mouse IgG1) or UPC10 (IgG2a) mAb. Cells were washed twice with PBS and resuspended in PBS with 1% autologous rat serum. One hundred microliters of goat-anti-mouse IgG-FITC (1:650 dilution; Tago, Burlingame, California) was then incubated with the cells for 45 minutes on ice. Cells were washed twice with PBS and fixed using a 1% paraformaldehyde solution. The samples were analyzed by FACScan (Beckton-Dickinson). Ten thousand events per sample were collected.
Antigen Proliferation Assays
EP-specific T lymphocyte cell lines and T cells recovered from cervical lymph nodes were cultured in triplicate in 0.2 ml of initiation medium at a density of 5 × 104 cells per well in 96-well U-bottom microtiter plates (Falcon, Lincoln Park, NJ). As APC, 2.5 × 105 irradiated (1,500 rad) mixed lymph node and thymus cells from Lewis rats were used. In specific experiments in which lymph node cells were recovered from TLD-4A2-treated animals, no accessory cells were added. EP peptide was added to the wells at 20 μg/ml final concentration. Forty-eight hours later, 0.5 μCi 3H-thymidine was added to each well of the plate. The cells were harvested 16 hours after the 3H-thymidine pulse (1295–001 Cell Harvester; LKB-Wallac, Gaithersburg, Maryland). Proliferation was measured by uptake of radioactivity and cpm were determined using liquid scintillation counting. For antibody blocking studies, 50 uL of OX-3 and OX-6 supernate or 1 to 10 μg of purified TLD-4A2, anti VLA-4, and anti ICAM-1 mAb were preincubated with APC for 60 minutes before the addition of T cells and antigen. In some wells, a combination of TLD-4A2, anti-ICAM-1, and anti-VLA-4 (10 μg/ml each) were used. Control wells consisting of APC, CD4+ T cells, and 50 μg/ml of mAb were also used to assess whether antibody alone might stimulate T cell proliferation.
Western Blot Studies
Lysates were prepared from either normal rat lymph-node cells or from monolayers of a rat SV40-transfected brain endothelial cell line designated GP8.E4. In each case the cells were surface-biotinylated according to the method of Cole et al (1987), with the following modification for the endothelium. Monolayers were washed six times before and after biotinylation, and sulfo-NHS-biotin (Pierce) in PBS was substituted for NHS-biotin in PBS/DMSO. Lysates from 1 × 107 cells were prepared in 2% NP-40, 10 mm Tris/HCL pH 8.0, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF. Precipitates were taken by mixing the lysates with 20 ml of protein-G sepharose with prebound antibody, for 1 hour at 4° C. Precipitates were washed successively in Tris/HCl containing 0.5 m NaCl, 1% NP40; 0.15% NaCl, 1% NP40; and finally 0.5% NP40, before boiling in SDS sample buffer. Precipitated proteins were separated in one- or two-dimensional SDS gels (non-reduced/reduced), (Male and Roitt, 1979), blotted onto nitrocellulose, and probed with 1:1,000 streptavidin-peroxidase (Amersham) in PBS, 5 mg/ml BSA, 0.1% Tween-20. The biotinylated proteins were visualized by enhanced chemiluminescence (ECL; Amersham) and autoradiography.
References
Aho R, Jalkanen S, and Kalimo H (1994). CD44-hyaluronate interaction mediates In vitro binding to the white matter of the central nervous system. J Neuropath Exp Neurol 53: 295–302.
Archelos J, Jung S, Maurer M, Schmied M, Lassmann H, Tamatani T, Miyasaka M, Toyka KV, and Hartung HP (1993). Inhibition of experimental autoimmune encephalomyelitis by an antibody to the intercellular adhesion molecule ICAM-1. Ann Neurol 34: 145–154.
Barclay AN, Beyers AD, Birkeland ML, Brown MH, Davis SJ, Somoza C, and Williams AF (1994). The leukocyte antigen facts book. San Diego, California: Academic Press.
Barker CF and Billingham RE (1977). Immunologically privileged sites. Adv Immunol 25: 1–6.
Baron JL, Madri JA, Ruddle NH, Hashim G, and Janeway CA (1993). Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med 177: 57–68.
Bauer J, Huitinga I, Zhao W, Hickey WF, and Dijkstra CD (1995). The role of macrophages, perivascular cells and microglial cells in the pathogenesis of experimental autoimmune encephalomyelitis. Glia 15: 437–446.
Bauer J, Sminia T, Wouterlood FG, and Dijkstra CD (1994). Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J Neurosci Res 38: 365–375.
Brosnan C, Bornstein MB, and Bloom B (1981). The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. J Immunol 126: 614–620.
Brostoff SW and Mason DW (1984). Experimental allergic encephalomyelitis: successful treatment in vivo with a monoclonal antibody that recognizes T helper cells. J Immunol 133: 1938–1942.
Cannella B, Cross AH, and Raine CS (1990). Upregulation and coexpression of adhesion molecules correlate with relapsing autoimmune demyelination in the central nervous system. J Exp Med 172: 1521–1524.
Cole SR, Ashman LK, and Ey PL (1987). Biotinylation: an alternative to radioiodination for the identification of surface antigens in immunoprecipitates. Mol Immunol 24: 699–705.
Cross AH, Cannella B, Brosnan CF, and Raine CS (1990). Homing to central nervous system vasculature by antigen-specific lymphocytes. I. Localization of 14 C-labeled cells during acute, chronic, and relapsing experimental allergic encephalomyelitis. Lab Invest 63: 162–170.
Cross AH, Girard TJ, Giacoletto KS, Evans RJ, Keeling RM, Lin RF, Trotter JL, and Karr RW (1995). Long-term inhibition of murine experimental autoimmune encephalomyelitis using CTLA-4-Fc supports a key role for CD28 costimulation. J Clin Invest 95: 2783–2789.
Damoiseaux JG, Dopp EA, Calame W, Chao D, MacPherson GC, and Dijkstra CD (1994). Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology 83: 140–147.
Flaris N, Densmore TL, Molleston MC, and Hickey WF (1993). Characterization of microglia and macrophages in the CNS of rats. Glia 7: 34–40.
Gerrotse K, Noelle RJ, Aruffo A, Ledbetter JA, Laman JD, Boersma JA, and Claassen E (1996). Functional and histological evidence for the involvement of gp39 (CD40 ligand) in multiple sclerosis. Proc Natl Acad Sci USA 93: 2499–2504.
Graeber MB, Streit WJ, Kiefer S, Schoen SW, and Kreutzberg GW (1990). New expression of myelomonocytic antigens by microglia and perivascular cells after lethal motor neuron injury. J Neuroimmunol 27: 121–126.
Graeber MB, Streit WJ, Sparks L, and Kreutzberg GW (1992). Ultrasturctural location of major histocompatibility complex (MHC) class II positive perivascular cells in histologically normal human brain. J Neuropath Exp Neurol 51: 303–311.
Hickey WF (1991). Migration of hematogenous cells through the blood brain barrier and the initiation of CNS inflammation. Brain Pathol 1: 97–102.
Hickey WF, Hsu BL, and Kimura H (1991). T-lymphocytes entry into the central nervous system. J Neurosci Res 28: 254–259.
Hickey WF and Kimura H (1988). Perivascular microglia are bone marrow derived and present antigen in vivo. Science 239: 290–292.
Hickey WF, Cohen JA, and Burns JB (1987). A quantitative immunohistochemical comparison of actively versus adoptively induced experimental allergic encephalomyelitis in the Lewis rat. Cell Immunol 109: 272–281.
Hickey WF, Gonatas NK, Kimura H, and Wilson DB (1983). Identification and quantification of T-lymphocyte subsets found in the spinal cord of Lewis rats during acute EAE. J Immunol 131: 2805–2809.
Huitinga I, Damoiseaux JG, Dopp EA, and Dijkstra CD (1993). Treatment with anti-CR3 antibodies ED7 and ED8 suppresses experimental allergic encephalomyelitis in Lewis rats. Eur J Immunol 23: 709–715.
Huitinga I, van Rooijen N, de Groot CJA, Uitdehaag BMJ, and Dijkstra CD (1990). Elimination of macrophages infiltrating the CNS suppresses EAE in rats. J Exp Med 172: 1025–10333.
Hulkower K, Brosnan CF, Aquino DA, Cammer W, Kulshrestha S, Guida MP, Rapoport DA, and Berman JW (1993). Expression of CF-1, C-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J Immunol 150: 2525–2533.
Jackson DG, Buckley J, and Bell JI (1992). Multiple variants of the human lymphocyte homing receptor CD44 generated by insertions at a single site in the extracellular domain. J of Biol Chem 267: 4732–4739.
Kent SJ, Karlik SJ, Cannon CC, Hines DK, Yednock TA, Fritz LC, and Horner HC (1995). A monoclonal antibody to alpha 4 integrin suppresses and reverses active experimental allergic encephalomyelitis. J Neuroimmunol 58: 1–10.
Kuchroo VK, Martin CA, Greer JM, Ju S-T, Sobel RA, and Dorf ME (1993). Cytokines and adhesion molecules contribute to the ability of myelin proteolipid protein-specific T cell clones to mediate experimental allergic encephalomyelitis. J Immunol 151: 4371–4382.
Larson RS and Springer TA (1990). Structure and function of leukocyte integrins. Immunol Rev 114: 181–217.
Lassmann H, Schmied M, Vass K, and Hickey WF (1991). Bone marrow derived elements and resident microglia in brain inflammation. Glia 7: 19–24.
Male D, Rahman J, Zhao W, and Hickey W (1995). An interferon-inducible molecule on brain endothelium which controls lymphocyte adhesion mediated by integrins. Immunol 64: 453–460.
Male DK and Roitt IM (1979). Analysis of the components of immune complexes. Mol Immunol 16: 197–203.
Mannie MD, Paterson PY, U’Prichard DC, and Flouret G (1985). Induction of EAE in Lewis rats with purified synthetic peptides: delineation of antigenic determinants for encephalitogenicity, In vitro activation of cellular transfer, and proliferation of lymphocytes. Proc Natl Acad Sci USA 82: 5519–5521.
Molleston MC, Thomas ML, and Hickey WF (1993). Novel MHC expression by microglia and site specific experimental allergic encephalomyelitis lesions in the rat after optic nerve transection. Adv Neurol 59: 337–348.
Moretto G, Xi RY, and Kim SU (1993). CD44 expression in human astrocytes and oligodendrocytes in culture. J Neuropath Exp Neurol 52: 419–423.
Pigott R and Power C (1994). The adhesion molecule facts book. Academic Press Inc, San Diego, California.
Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, and Tuohy VK (1993). Astrocyte expression of mRNA encoding cytokines IP-20 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB 7: 592–600.
Shimizu Y and Shaw S (1991). Lymphocyte interactions with extracellular matrix. FASEB 5: 2292–2300.
Sloan DJ, Wood MJ, and Charlton HM (1992). Leucocyte recruitment and inflammation in the CNS. TINS 15: 276–279.
Stockinger H, Gadd SJ, Eher R, Majdic O, Schreiber W, Kasinrerk W, Strass B, and Knapp W (1990). Molecular characterization and functional analysis of leukocyte surface protein CD31. J Immunol 145: 3889–3897.
Sriram S and Roberts CA (1986). Treatment of established chronic relapsing experimental allergic encephalomyelitis either anti-L3T4 antibodies. J Immunol 86: 4464–4469.
Sriram S and Steinman L (1983). Anti I-A antibody suppresses active encephalomyelitis: treatment model for diseases linked to IR genes. J Exp Med 158: 1362–1367.
Steinman L, Rosenbaum JT, Sriram S, and McDevitt HO (1981). in vivo effects of antibodies to immune response gene products: prevention of experimental allergic encephalitis. Proc Natl Acad Sci USA 78: 7111–7114.
Unger ER, Sung JH, Manivel JC, Chenggis ML, and Blazar BR (1993). Male donor derived cells in the brains of female sex-mismatched bone marrow transplant recipients: a Y-chromosome specific in situ hybridization study. J Neuropath Exp Neurol 52: 460–470.
Wekerle H, Linington H, Lassmann H, and Meyermann R (1986). Cellular immune reactivity within the CNS. Trends Neurosci 9: 271–273.
Williams K, Bar-Or A, Ulvestad E, Olivier A, Antel JP, and Yong VW (1992). Biology of adult human microglia in culture: comparisons with peripheral blood monocytes and astrocytes. J Neuropath Exp Neurol 51: 538–549.
Williams K, Ulvestad E, and Antel JP (1994a). B7/BB-1 antigen expression on adult human microglia studied In vitro and in situ. Eur J Immunol 24: 3031–3037.
Williams KC, Ulvestad E, and Hickey WF (1994b). Immunology of multiple sclerosis. Clin Neurosci 2: 229–245.
Williams KC, Zhao W, Uneo K, and Hickey WF (1996). Treatment of Lewis rats with 3A12 mAb against PECAM-1 the role of PECAM in EAE induction. J Neurosci Res 45: 747–757.
Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, and Karin N (1992). Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 356: 63–66.
Zaller DM, Osman G, Kanagawa O, and Hood L (1990). Prevention and treatment of murine experimental allergic encephalomyelitis with T cell receptor v-beta specific antibodies. J Exp Med 171: 1943–1955.
Acknowledgements
We thank Dr. A.A. Lackner and Dr. A. McClean for critical review of the manuscript, and Ms. K. Toohey for photographic and graphic services.
This work was supported in part by grants RG 2856-A-1 (KCW), NS-37654 (KCW) and NS-27321 (WFH).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Williams, K., Zhao, W., Politopoulou, G. et al. Inhibition of Experimental Allergic Encephalomyelitis with an Antibody that Recognizes a Novel Antigen Expressed on Lymphocytes, Endothelial Cells, and Microglia. Lab Invest 80, 313–326 (2000). https://doi.org/10.1038/labinvest.3780036
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780036
This article is cited by
-
Angiogenesis is present in experimental autoimmune encephalomyelitis and pro-angiogenic factors are increased in multiple sclerosis lesions
Journal of Neuroinflammation (2010)
-
Distribution and immunoregulatory properties of antisecretory factor
Laboratory Investigation (2004)
