
Abstract
Caveolins are the principal protein components of caveolae, invaginations of the plasma membrane involved in cell signaling and trafficking. Caveolin-3 (Cav-3) is the muscle-specific isoform of the caveolin family and mutations in the CAV3 gene lead to a large group of neuromuscular disorders. In unrelated patients, we identified two distinct CAV3 mutations involving the same codon 78. Patient 1, affected by dilated cardiomyopathy and limb girdle muscular dystrophy (LGMD)-1C, shows an autosomal recessive mutation converting threonine to methionine (T78M). Patient 2, affected by isolated familiar hyperCKemia, shows an autosomal dominant mutation converting threonine to lysine (T78K). Cav-3 wild type (WT) and Cav-3 mutations were transiently transfected into Cos-7 cells. Cav-3 WT and Cav-3 T78M mutant localized at the plasma membrane, whereas Cav-3 T78K was retained in a perinuclear compartment. Cav-3 T78K expression was decreased by 87% when compared with Cav-3 WT, whereas Cav-3 T78M protein levels were unchanged. To evaluate whether Cav-3 T78K and Cav-3 T78M mutants behaved with a dominant negative pattern, Cos-7 cells were cotransfected with green fluorescent protein (GFP)-Cav-3 WT in combination with either mutant or WT Cav-3. When cotransfected with Cav-3 WT or Cav-3 T78M, GFP-Cav-3 WT was localized at the plasma membrane, as expected. However, when cotransfected with Cav-3 T78K, GFP-Cav-3 WT was retained in a perinuclear compartment, and its protein levels were reduced by 60%, suggesting a dominant negative action. Accordingly, Cav-3 protein levels in muscles from a biopsy of patient 2 (T78K mutation) were reduced by 80%. In conclusion, CAV3 T78M and T78K mutations lead to distinct disorders showing different clinical features and inheritance, and displaying distinct phenotypes in vitro.
Similar content being viewed by others


Main
Caveolae are vesicular invaginations of the plasma membrane that regulate endocytosis, cholesterol homeostasis and signal transduction processes. Caveolins, 21–24 kDa integral membrane proteins, are the principal components of caveolae membranes and are part of a family of three isoforms encoded by different genes. Caveolin-1 (Cav-1) and -2 (Cav-2) are coexpressed in most cell types, whereas caveolin-3 (Cav-3) is synthesized specifically in both cardiac and skeletal muscle as well as smooth muscle cells.1
The human Cav-3 protein is 150 amino acids in length and is divided into four separate domains: N-terminal (aa 1–53), scaffolding (aa 54–73), transmembrane (aa 74–106) and C-terminal (aa 107–151).2, 3, 4
In the endoplasmic reticulum, Cav-3 initiates the biogenesis of caveolae organelles by forming homo-oligomers and hetero-oligomers with Cav-1.5 At the plasmalemma, Cav-3 interacts with dystrophin and its associated glycoproteins.6, 7 Cav-3 and dystrophin competitively bind to the same site of β-dystroglycan, suggesting that Cav-3 may regulate the membrane recruitment of dystrophin and the assembly of the dystrophin glycoprotein complex (DGC).8 At the cell surface, Cav-3 colocalizes also with signaling molecules such as Gi2α, Gβγ, c-Src, other Src kinases and nitric oxide synthases (neuronal and inducible NOS), indicating that muscle caveolae might be involved in the modulation of these signaling processes.9, 10, 11, 12 In addition, Cav-3 plays a role in the regulation of energy metabolism of muscle cells as it is required for the cell membrane targeting of phosphofructokinase, an enzyme that catalyzes a rate-limiting reaction in glycolysis.13, 14
In vitro studies have shown that Cav-3 plays a critical role in myoblast cell differentiation and survival and in myotube formation.15 The relevance of Cav-3 in muscle physiology was further confirmed by the findings that mutations in the CAV3 gene result in distinct neuromuscular and cardiac disorders, such as limb girdle muscular dystrophy (LGMD) 1-C, idiopathic persistent elevation of serum creatine kinase (hyperCKemia), inherited rippling muscle disease (RMD), distal myopathy and familial hypertrophic cardiomyopathy (HCM).16, 17, 18, 19, 20
The CAV3 gene (OMIM no. 601253) spans 12 kb of genomic DNA on chromosome 3p25 and contains two exons. At present, 20 different point mutations, 2 base-pair deletions and 1 novel splice site mutation have been reported.21 More recently, four novel CAV3 mutations have been identified in patients affected by congenital long-QT syndrome (LQTS) in the absence of signs of primary cardiomyopathy, suggesting a possible role for Cav-3 in the regulation of cardiac ion channels.21, 22
Functional analysis of different CAV3 missense mutations revealed that Cav-3 mutant proteins can be retained in the Golgi apparatus, where they are rapidly degraded by the ubiquitin–proteasome system and display a dominant negative effect on the wild-type (WT) protein.23, 24
In this study, we showed that two distinct CAV3 missense mutations affecting amino acid 78 cause different phenotypes in vivo and in vitro.
MATERIALS AND METHODS
Patients
Patient 1, a 58-year-old woman, reported myalgias, premature fatigue and dyspnea during moderate exercise. Blood samples for genetic analysis were collected after informed consent. The patient always declined to perform a muscle biopsy.
Patient 2, a 40-year-old man, was referred because of hyperCKemia and calf hypertrophy. Blood samples for genetic analysis and muscle biopsy for immunohistochemistry were collected for diagnostic purposes after informed consent and were processed according to standard histological and histochemical techniques. All the procedures were performed in accordance with Gaslini and Meyer Institute guidelines.
Genetic Analysis
Genomic DNA was extracted from patient's peripheral blood leukocytes using the FlexiGene DNA kit (Qiagen, Milan, Italy) following the manufacturer's instructions. The DNA was amplified by polymerase chain reaction (PCR) using the following primers: exon 1 forward 5′-gctggcagggacataagtc-3′, exon 1 reverse 5′-ctgtgtctgcagcagatac-3′; exon 2 forward 5′-aatacaggtagggtccagccac-3′, exon 2 reverse 5′-ggtatggagcagtccctaaagag-3′. PCR conditions were as follows: denaturation at 94°C for 4 min, 35 cycles of 94°C for 30 s, 58°C for 30 s, 72°C for 1 min and final extension at 72°C for 10 min.16 PCR products were purified with the Montage PCRμ96 Purification Kit (Millipore Corporation, Bedford, MA, USA) and sequenced on both strands using the ABI Prism Big Dye Terminator Cycle Sequencing Kit and an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The mutations identified by direct sequencing were confirmed by specific PCR restriction fragment length polymorphism (RFLP) (data not shown). To exclude the possibility of polymorphism variants, 200 DNA controls were sequenced using the same experimental design. Genetic analysis of the LMNA gene was performed as previously described.25
cDNA Constructs
Full-length WT Cav-3 was subcloned into the vector pCAGGS.23 Standard PCR strategies were used to construct the various point mutants.23 The rat Cav-3 WT cDNA was used as the template for these reactions. The various Cav-3 point mutants were subcloned into the pCAGGS vector using the EcoRI sites within the polylinker. Correct orientation was verified by protein expression. DNA sequencing confirmed the accuracy of the intended mutations.
Cells, Media and Transfection Methods
Cos-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. Cos-7 cells were transfected using the Effectene transfection reagent (Qiagen). At 36 or 48 h after transfection, cells were harvested.
Histological and Histochemical Analysis
Unfixed 5-μm-thick cryosections of skeletal muscle biopsies from patient 2 were subjected to hematoxylin and eosin (H&E) staining, as previously described.16
Immunohistochemistry
Unfixed 5-μm-thick cryosections of skeletal muscle biopsies from patient 2 were incubated for 1 h at room temperature (RT) with a 1:800 mouse monoclonal anti-Cav-3 antibody (anti-Cav-3 mAb, clone 26 raised against amino-acid residues 3–24; Transduction Laboratories, Lexington, KY, USA) diluted in phosphate-buffered saline (PBS)/1% bovine serum albumin (BSA), with an undiluted mouse monoclonal anti-dysferlin antibody (Hamlet II, Novocastra Laboratories, Newcastle upon Tyne, UK), with an undiluted mouse monoclonal anti-dystrophin (N-terminus) antibody (NCL-DYS3, Novocastra Laboratories) or with an undiluted mouse monoclonal anti-α-sarcoglycan antibody (Novocastra Laboratories). After repeated washes with PBS, sections were incubated for 1 h at RT with a 1:100 biotinylated anti-mouse IgG (Amersham Biosciences, Little Chalfont, UK) and exposed for 30 min at RT to a 1:250 streptavidin fluorescein (Amersham Biosciences) in the dark. Finally, the sections were mounted with glycerol (glycerol 87% diluted 2:1 in PBS) and observed under a Leica Diaplan microscope.26
Western Immunoblot Analysis
Skeletal muscle biopsy
Five to eight 10-μm-thick cryosections of skeletal muscle biopsy were dissolved in lysis buffer (4% sodium dodecyl sulfate (SDS) in 4 M urea). Tissue lysates were then sonicated for 10 s in ice and centrifuged at 10 000 r.p.m. for 15 min at 4°C. Supernatants were collected and protein concentration was determined by using the Lowry protein assay method. Equal amounts of protein lysates were resolved by SDS-PAGE (12% acrylamide) under reducing conditions and transferred to nitrocellulose membranes (Immobilon Plus, Millipore, Billerica, MA, USA). The proteins were visualized with Ponceau S (Sigma, Milan, Italy). Membranes were blocked with 4% non-fat dried milk in PBST (PBS/0.1% Tween 20). Blots were then incubated for 2 h at RT with a 1:1000 anti-Cav-3 antibody, a 1:300 anti-dysferlin antibody or a 1:50 mouse monoclonal antibody against vinculin (Vinculin, Novocastra), all diluted in PBS/1% BSA. Horseradish peroxidase-conjugated anti-mouse IgG was used to visualize bound primary antibodies with the ECL chemiluminescence system (Amersham Biosciences). Band intensities were evaluated by densitometry using the Molecular Imager ChemiDoc XRS system (Biorad Laboratories, Hercules, CA, USA).
Cos-7 cells
Cos-7 cells were transiently transfected with WT or mutant forms of Cav-3. Cells were lysed in boiling sample buffer. Samples were collected, homogenized using a 26 G needle and 1 ml syringe and boiled for a total of 5 min. Equal amounts of the protein lysates were resolved by SDS-PAGE (12% polyacrylamide) under reducing conditions and transferred to nitrocellulose membranes.
Membranes were blocked with 4% non-fat dried milk in TBST (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20). Blots were then incubated for 1 h at RT with a 1:1000 anti-Cav-3 antibody, a 1:1000 mouse polyclonal anti-green fluorescent protein (GFP) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or a 1:1000 monoclonal anti-GAPDH antibody (RDI, Flanders, NJ, USA) all diluted in TBST/1% BSA.
Horseradish peroxidase-conjugated anti-mouse IgG antibodies were used to visualize bound primary antibodies with the supersignal chemiluminescence substrate (Pierce, Rockford, IL, USA).
Immunofluorescence Analysis
Transfected Cos-7 cells were fixed for 30 min in PBS containing 2% paraformaldehyde. After fixation, cells were rinsed with PBS and permealized with IF buffer (PBS plus 0.2% BSA and 0.1% Triton X-100) for 10 min. Then, cells were treated with NH4Cl in PBS for 10 min to quench free aldehyde groups. Primary antibodies (anti-Cav-3 and anti-GFP) were incubated in IF buffer for 1 h at RT. After washing with PBS, the bound primary antibody was visualized with a tagged secondary antibody (Jackson Immunoresearch, Newmarket, UK). Slides were mounted with slow-fade anti-fade reagent (Molecular Probes, Eugene, OR, USA) and observed with a confocal microscope.
RESULTS
Two patients presenting with signs and symptoms suggestive of primitive myopathies such as myalgias, muscle weakness, calf hypertrophy or isolated hyperCKemia were screened for CAV3 mutations.
Patient 1, a 58-year-old woman born to consanguineous parents, came to our observation because of myalgias, premature fatigue and dyspnea during moderate exercise. At 47 years, she had been diagnosed with dilated cardiomyopathy (DCM). Family history was negative for neuromuscular disorders. Neurological examination revealed generalized hypotonia, proximal weakness and hypotrophy of the upper limbs, and proximal hypotrophy and calf hypetrophy of the lower limbs. Consistent with a myopathic pattern, electromyography of lower limbs showed decreased amplitude and duration of response, and signs of increased spontaneous activity, such as fibrillations and positive sharp waves. Laboratory investigations revealed increased serum creatine kinase (CK) levels (>1500 IU/l; normal value <150). Serum lactic acid, urinary organic acids and acylcarnitine profile were normal. Echocardiographic evaluation evidenced left ventricular dilation with severe reduction of the ejection fraction (about 16%), whereas the electrocardiograms (basal and Holter) revealed an incomplete left bundle branch block but no arrhythmias. She refused muscle biopsy.
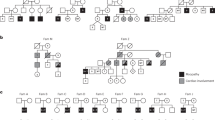
Genetic analysis of the CAV3 gene identified a homozygous mutation at nucleotide 233 (C>T) leading to an amino-acid substitution, threonine to methionine, at codon 78 (T78M). Her parents (deceased before the study was undertaken) did not report any clinical symptom suggestive of the disease. The patient's daughter, heterozygous for the mutation, did not show any symptoms of Cav-3 deficiency and her serum CK levels were normal. Taken together, these data indicate an autosomal recessive mode of inheritance for this trait (Figure 1a). Laminopathies were ruled out by sequencing the LMNA gene.

Genetic analysis of CAV3 in two pedigrees. (a) Electropherograms showing WT sequence and two different missense mutations affecting codon 78 of CAV3. (b) Pedigrees of the families. Arrows indicate the probands, + the WT allele, − the mutated allele, partially filled symbols individuals affected by hyperCKemia, filled symbol patient 2 affected by LGMD1-C and DCM and open symbols unaffected individuals.
Patient 2, a 40-year-old man, was referred because of hyperCKemia and calf hypertrophy. Family history was positive for hyperCKemia. He did not describe myalgias, exercise intolerance or progressive muscle weakness, and neurological examinations revealed no abnormalities. Electrocardiography was normal and cardiovascular evaluation did not show any alterations.
Electromyography of upper and lower limbs displayed a myopathic pattern. Laboratory investigations revealed increased serum CK levels (900 IU/l, normal value <150 IU/l). Genetic analysis of the CAV3 gene evidenced a novel heterozygous mutation at nucleotide position 233 (C>A) converting threonine to lysine at codon 78 (T78K). This single base change was found in I:1, III:2 and III:3, who are affected by isolated hyperCKemia, and was not detected in the unaffected member III:1, in accordance with an autosomal dominant mode of transmission (Figure 1b).
Immunohistochemical studies of the muscle biopsy revealed that Cav-3 expression at the cell surface was markedly reduced. This effect was associated with a decreased membrane staining of dysferlin (Figure 2a). The defect of Cav-3 and dysferlin was not due to nonspecific membrane damage. Indeed, the morphological analysis of the biopsy did not show myopathic signs such as variability in muscle fiber size, degenerating/regenerating fibers with an increased number of central nuclei and increase in connective tissue. Moreover, immunostaining for dystrophin and α-sarcoglycan, two critical muscle membrane proteins, was normal (Figure 2b).

CAV3 T78K mutation in patient 2 causes a severe decrease in Cav-3 expression and alters dysferlin membrane localization. (a) Frozen sections of the skeletal muscle biopsy from patient 2 (Pt. 2) and an age-matched control (Control) were prepared and immunostained with specific antibodies against caveolin-3 (Cav-3) and dysferlin (Dysf). In control muscle, Cav-3 and Dysf display a uniform pattern at the sarcolemma. In patient 2, Cav-3 is markedly reduced, whereas Dysf is only partially decreased and characterized by a disorganized patchy staining at the cell surface. Final magnification, × 40. Size bars correspond to 50 μm. (b) Frozen sections of the skeletal muscle biopsy from patient 2 (Pt. 2) were analyzed with H&E staining and subjected to immunohistochemistry with specific antibodies against dystrophin (N-terminus) and α-sarcoglycan (α-Sarcogl.) No myopathic changes such as variability in fiber size, degenerating/regenerating muscle fibers with an increased number of central nuclei and increase in connective tissue were detected. Dystrophin and α-Sarcogl. immunostaining was normal. Final magnification, × 40. Size bars correspond to 50 μm.
The reduction of Cav-3 protein levels (approximately 80%) was confirmed by immunoblot analysis on total muscle lysate (Figure 3).

CAV3 T78K mutation in patient 2 causes a decrease in Cav-3 expression. Protein lysates prepared from the skeletal muscle biopsy from patient 2 (Pt. 2) and an age-matched control (C) were separated by SDS-PAGE and subjected to western immunoblot analysis using antibodies against Cav-3. In patient 2, Cav-3 protein levels were reduced by approximately 80%. Equal loading control was assessed by immunoblot with antibodies against vinculin (Vinc).
To investigate the impact of T78M and T78K mutations on Cav-3 protein levels and cellular localization, we generated Cav-3 T78M and T78K mutants and cloned them into a mammalian expression vector. We next transfected Cos-7 cells with these mutants and assessed their expression relative to Cav-3 WT protein by immunoblot analysis using a specific Cav-3 antibody. Cav-3 T78K was markedly reduced (about 87%) when compared with the WT construct, whereas Cav-3 T78M protein levels were not changed (Figure 4). Immunofluorescence analysis revealed that Cav-3 T78K was retained intracellularly in a perinuclear compartment. In contrast, Cav-3 WT and Cav-3 T78M were correctly targeted to the plasma membrane (Figure 5).

Cav-3 T78K mutant is expressed at lower levels. Cos-7 cells were transiently transfected with cDNAs encoding Cav-3 WT, Cav-3 T78M or Cav-3 T78K. At 36 h after transfection, cells were extracted in lysis buffer. Protein lysates were separated by SDS-PAGE and subjected to immunoblot analysis with antibodies against Cav-3. Cav-3 T78K is expressed at much lower levels than Cav-3 WT. Cav-3 T78M expression levels are unchanged. Equal loading was assessed by immunoblot with antibodies against glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Immunoblots are representative of three independent cultures.

Cav-3 T78K mutant is retained in an intracellular compartment. Cos-7 cells were transiently transfected with cDNA encoding Cav-3 WT, Cav-3 T78M or Cav-3 T78K. At 36 h after transfection, cells were fixed and stained with antibodies against Cav-3. Cav-3 WT is localized at the plasma membrane, as expected. Cav-3 T78M mutant does not change the cellular distribution. However, Cav-3 T78K is retained in an intracellular, perinuclear compartment. N, nucleus. The data shown are representative of three independent cultures. Final magnification, × 100. Size bars correspond to 20 μm.
We next examined whether Cav-3 T78K and Cav-3 T78M had a dominant negative effect as described for LGMD-1C Cav-3 mutations.23 For this purpose, the two mutants were cotransfected with an expression vector containing Cav-3 WT fused in-frame with a GFP cDNA (GFP-Cav-3 WT). At 36 and 48 h after transfection, cells were fixed and stained with antibodies against GFP. In this way, we could follow the behavior of GFP-Cav-3 WT when expressed along with the Cav-3 WT and mutant forms. When cotransfected with Cav-3 WT or Cav-3 T78M, GFP-Cav-3 WT was localized at the plasma membrane, as expected. However, when cotransfected with Cav-3 T78K, GFP-Cav-3 WT was retained in a perinuclear compartment (Figure 6). In addition, immunoblot analysis using an antibody directed against GFP indicated that, in the presence of Cav-3 T78M, GFP Cav-3 WT expression was not affected, whereas when cotransfected with Cav-3 T78K, its protein levels were decreased by 36%, suggesting that the T78K mutation can induce degradation of the WT form. This effect increased over time, and at 48 h following transfection, GFP-Cav-3 WT protein was reduced by 60% (Figure 7).

Cav-3 T78K mutant causes intracellular retention of Cav-3 WT form. Cos-7 cells were transiently cotransfected with cDNA encoding GFP-Cav-3 WT in combination with Cav-3 WT, Cav-3 T78M or Cav-3 T78K. At 36 and 48 h after transfection, cells were fixed and stained with antibodies against GFP. When cotransfected with Cav-3 WT or Cav-3 T78M, GFP-Cav-3 WT is localized at the plasma membrane, as expected. When cotransfected with Cav-3 T78K, GFP-Cav-3 WT is retained in a perinuclear compartment, suggesting that Cav-3 T78K behaves in a dominant negative manner. N, nucleus. The data shown are representative of four independent cultures. Final magnification, × 100. Size bars correspond to 20 μm.

Cav-3 T78K affects the expression levels of Cav-3 WT while Cav-3 T78M does not display a dominant negative effect. (a) Cos-7 cells were transiently cotransfected with cDNAs encoding GFP-Cav-3 WT in combination with Cav-3 WT, Cav-3 T78M or Cav-3 T78K. At 36 and 48 h after transfection, cells were extracted in lysis buffer. Protein lysates were separated by SDS-PAGE and subjected to immunoblot analysis with antibodies against GFP. When cotransfected with Cav-3 WT and Cav-3 T78M, GFP Cav-3 WT is expressed at similar levels, suggesting that Cav-3 T78M mutant does not induce degradation of the WT form. In the presence of Cav-3 T78K, GFP Cav-3 WT protein levels are reduced by approximately 30% at 36 h and by 60% at 48h following transfection. Equal loading was assessed by immunoblot with antibodies against GAPDH. (b) Experiments are depicted as a graph. Immunoblots are representative of two cultures.
DISCUSSION
In this study, we describe two distinct CAV3 missense mutations that, although occurring at the same codon 78, lead to different clinical phenotypes.
Patient 1, who carries an autosomal recessive CAV3 T78M amino-acid substitution, is affected by DCM and LGMD-1C.
DCM is a common clinical feature in different neuromuscular pathologies, but it has never been described in a caveolinopathy.20, 27, 28, 29, 30 The clinical examination, laboratory tests and genetic analyses excluded the coexistence of other disorders including laminopathy or a mitochondrial disease. However, the lack of a muscle biopsy, due to refusal by the patient, does not allow us to conclude unequivocally that her cardiac phenotype is due to the CAV3 mutation. It could also be possible that our patient's cardiomyopathy was not primarily dilatative but represented the late clinical course of a hypertrophic condition. This would be in agreement with the description by Hayashi et al,31 who reported the occurrence of HCM in two brothers affected by a CAV3 missense variant.
Indeed, the role of CAV3 mutations in the pathogenesis of cardiac disorders is complex and has not yet been clarified.
Cav-3 is expressed in cardiomyocytes, where it colocalizes with different molecules involved in the development of myocardium hypertrophy, such as heterotrimeric G proteins, protein kinase C (PKC), Ras, extracellular signal-regulated protein kinase (ERK) and cNOS. As Cav-3 inhibits the activation of these signaling pathways, it could be hypothesized that caveolae might function as a control center of cellular signaling during cardiomyocyte hypertrophy.32 Consistent with these data, in rats, overexpression of Cav-3 inhibits agonist-induced cardiomyocyte hypetrophy, whereas the dominant negative Cav-3 enhances this process.33 In accordance with this, transgenic mice overexpressing the Cav-3 P104L mutant develop the unique pathophysiological characteristics of HCM, such as increased thickness of the interventricular septum and the left ventricular posterior wall, hypercontractility and diastolic dysfunction.34 Finally, Cav-3-knockout mice display eccentric left ventricular hypertrophy, which then progresses to chamber dilation and 20% reduced fractional shortening.35
In humans, the vast majority of CAV3 mutations do not cause cardiac phenotypes and the two single reports available in the literature are controversial. On the one hand, as mentioned earlier, one study describes the occurrence of HCM in two brothers carrying a CAV3 T63S substitution.31 On the other hand, Cav-3 expression and caveolar structures were normal in the cardiac muscle tissue of a patient affected by hyperCKemia and RMD associated with a severe Cav-3 deficit in skeletal muscle.36 These data seem to suggest different mechanisms of regulation of the Cav-3 protein in the skeletal and cardiac muscle tissues and are suggestive of future studies.
It is important to highlight that the CAV3 T78M amino-acid change was described in the heterozygous state in four subjects affected by congenital LQTS.22 These patients did not exhibit signs of skeletal muscle involvement or primary cardiomyopathy, thus implicating a possible role for Cav-3 and caveolae in cardiac excitability. This is not surprising as β2-adrenoreceptors colocalize with Cav-3 in ventricular and sinoatrial myocytes, and chemical disruption of caveolae affects the excitation–contraction coupling and β-adrenergic responsiveness of adult cardiac myocytes.37, 38, 39 In this study, we did not detect any signs of arrhythmia in our patient or in her heterozygous daughter.
In another recent study, a heterozygous CAV3 T78M mutation was detected in one patient affected by idiopathic hyperCKemia.40 However, in our report, the heterozygous daughter of patient 1 displayed normal serum CK levels.
The CAV3 T78K variant is novel and was identified in a family with autosomal dominant isolated hyperCKemia. Analysis of the muscular biopsy from the proband revealed a severe reduction in Cav-3 expression. Analogous with what was observed with the Cav-3 mutants, R26Q or P104L, the decrease of Cav-3 protein was associated with a reduction of the plasma membrane localization of dysferlin, a 230 kDa transmembrane protein, which plays a role in membrane repair processes and whose mutations lead to LGMD type 2B, Miyoshi myopathy and distal anterior compartment myopathy.41, 42, 43 Cav-3 and dysferlin colocalize at the sarcolemma of mature muscle fibers and the Cav-3 T78K mutation inhibits dysferlin expression at the plasmalemma.
Because the clinical phenotypes of patients 1 and 2 are different, we hypothesized that the functional alterations due to the distinct mutations might be qualitatively or quantitatively distinct. To test this hypothesis, we analyzed the phenotypic behavior of these mutants in a heterologous system using Cav-3 T78M and T78K constructs. In vitro characterization of the Cav-3 T78M mutant revealed that this mutation does not affect Cav-3 protein expression and membrane localization, and, therefore, appears to act through a loss-of-function mechanism of action. These results might explain the manifestation of a severe muscular phenotype in the patient carrying the CAV3 T78M substitution in the homozygous state.
In contrast, Cav-3 T78K markedly decreases the steady-state expression levels of Cav-3 and leads to its intracellular retention in the Golgi complex. Moreover, Cav-3 T78K mutant behaves in a dominant negative fashion, causing the trapping of WT Cav-3 within the Golgi and inducing its degradation. Although the cellular behavior of this mutant is consistent with its autosomal dominant inheritance, these in vitro data seem to be in conflict with the mild phenotype displayed by the patient, that is, isolated hyperCKemia, and no myopathic changes on histopathological examination. In reality, in caveolinopathies, the genotype–phenotype correlation is not always preserved. Other genetic modifiers may be implied in determining the phenotypic presentation of Cav-3 deficiency, so that the same CAV3 mutation can lead to heterogeneous clinical phenotypes and muscle histopathological changes.21, 44 Indeed, the same R26Q CAV3 mutation was reported in a family with phenotypic variability. Of the 11 individuals with the confirmed mutation, 3 exhibited both RMD and LGMD-1C features, 2 had predominantly LGMD-1C features, 2 had minor muscle stiffness only and 1 was asymptomatic.45 This observed intrafamilial and interfamilial phenotypic variability suggests the involvement of modifying genetic background effects on the development of the patients' phenotype.
Threonine 78 is an evolutionarily highly conserved residue and is located in the Cav-3 hydrophobic domain (residues 74–106). This region spans the membrane and forms a hairpin loop within the sarcolemma, thus allowing both the N- and C-terminal segments to face the cytoplasm.46 Studies on caveolin membrane-spanning segments showed that this domain plays a critical role in the genesis of those Cav-3 homo-oligomers that are thought to represent the assembly units driving the formation of caveolae organelles in muscle cells.47, 48 We have suggested that the substitution of the neutral residue threonine 78 with the positively charged lysine causes misfolding of Cav-3 protein impairing the process of Cav-3 self-association and leading to Cav-3 rapid degradation. On the other hand, we can speculate that the introduction of a neutral methionine residue in the same position results in a stable but dysfunctional Cav-3 protein. Future studies will address the effects of Cav-3 T78M on candidate signaling pathways known to interact with the Cav-3 protein, especially in the context of heart-derived cells.49
In conclusion, CAV3 T78M and T78K mutations correlate with distinct disorders showing different clinical features and modes of inheritance. Consistently, they display a distinct phenotype in vitro. In addition, it is noteworthy to highlight that the CAV3 T78M mutation is possibly associated with the development of a cardiac phenotype. There is a growing body of evidence that CAV3 missense mutations may lead to a cardiac involvement, including cardiomyopathy and arrhythmia.
Extensive clinical examination of cardiac function in LGMD-1C, RMD and hyperCKemic CAV3 patients and mutational screening of large populations will be necessary to assess the role of CAV3 mutations in heart disease. In addition, the function of Cav-3 in the heart should be evaluated to unveil the pathogenetic mechanisms behind the cardiac clinical features of caveolinopathies.
References
Williams TM, Lisanti MP . The Caveolin genes: from cell biology to medicine. Ann Med 2004;36:584–595.
Tang Z, Scherer PE, Okamoto T, et al. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem 1996;271:2255–2261.
Scherer PE, Okamoto T, Chun M, et al. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci USA 1996;93:131–135.
Okamoto T, Schlegel A, Scherer PE, et al. Caveolins, a family of scaffolding proteins for organizing ‘preassembled signaling complexes’ at the plasma membrane. J Biol Chem 1998;273:5419–5422.
Capozza F, Cohen AW, Cheung MW, et al. Muscle-specific interaction of caveolin isoforms: differential complex formation between caveolins in fibroblastic vs. muscle cells. Am J Physiol Cell Physiol 2005;288:C677–C691.
Song KS, Scherer PE, Tang Z, et al. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem 1996;271:15160–15165.
Crosbie RH, Yamada H, Venzke DP, et al. Caveolin-3 is not an integral component of the dystrophin glycoprotein complex. FEBS Lett 1998;427:279–282.
Sotgia F, Lee JK, Das K, et al. Caveolin-3 directly interacts with the C-terminal tail of beta—dystroglycan. Identification of a central WW-like domain within caveolin family members. J Biol Chem 2000;275:38048–38058.
Li S, Okamoto T, Chun M, et al. Evidence for a regulated interaction between heterotrimeric G proteins and caveolin. J Biol Chem 1995;270:15693–15701.
Garcia-Cardena G, Fan R, Stern DF, et al. Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J Biol Chem 1996;271:27237–27240.
Shaul PW, Smart EJ, Robinson LJ, et al. Acylation targets emdothelial nitric-oxide synthase to plasmalemmal caveolae. J Biol Chem 1996;271:6518–6522.
Smythe GM, Eby JC, Disatnik MH, et al. A caveolin-3 mutant that causes limb girdle muscular dystrophy type 1C disrupts Src localization and activity and induces apoptosis in skeletal myotubes. J Cell Sci 2003;116 (Part 23):4739–4749.
Sotgia F, Bonuccelli G, Minetti C, et al. Phosphofructokinase muscle-specific isoform requires caveolin-3 expression for plasma membrane recruitment and caveolar targeting: implications for the pathogenesis of caveolin-related muscle diseases. Am J Pathol 2003;163:2619–2634.
Karlsson M, Thorn H, Parpal S, et al. Insulin induces translocation of glucose transporter GLUT4 to plasma membrane caveolae in adipocytes. FASEB J 2002;16:249–251.
Galbiati F, Volonte D, Engelman JA, et al. Targeted down-regulation of caveolin-3 is sufficient to inhibit myotube formation in differentiating C2C12 myoblasts. Transient activation of p38 mitogen-activated protein kinase is required for induction of caveolin-3 expression and subsequent myotube formation. J Biol Chem 1999;274:30315–30321.
Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 1998;18:365–368.
Merlini L, Carbone I, Capanni C, et al. Familial isolated hyperCKaemia associated with a new mutation in the caveolin-3 (CAV-3) gene. J Neurol Neurosurg Psychiatry 2002;73:65–67.
Carbone I, Bruno C, Sotgia F, et al. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology 2000;54:1373–1376.
Galbiati F, Razani B, Lisanti MP . Caveolae and caveolin-3 in muscular dystrophy. Trends Mol Med 2001;7:435–441.
Tsubata S, Bowles KR, Vatta M, et al. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest 2000;106:655–662.
Fulizio L, Nascimbeni AC, Fanin M, et al. Molecular and muscle pathology in a series of caveolinopathy patients. Hum Mutat 2005;25:82–89.
Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 2006;114:2104–2112.
Galbiati F, Volonte D, Minetti C, et al. Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). Retention of LGMD-1C caveolin-3 mutants within the golgi complex. J Biol Chem 1999;274:25632–25641.
Galbiati F, Volonte D, Minetti C, et al. Limb-girdle muscular dystrophy (LGMD-1C) mutants of caveolin-3 undergo ubiquitination and proteasomal degradation. Treatment with proteasomal inhibitors blocks the dominant negative effect of LGMD-1C mutanta and rescues wild-type caveolin-3. J Biol Chem 2000;275:37702–37711.
Moulson CL, Fong LG, Gardner JM, et al. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum Mutat 2007;28:882–889.
Minetti C, Bado M, Broda P, et al. Impairment of caveolae formation and T-system disorganization in human muscular dystrophy with caveolin-3 deficiency. Am J Pathol 2002;160:265–270.
Worman HJ, Bonne G . ‘Laminopathies’ a wide spectrum of human diseases. Exp Cell Res 2007;313:2121–2133.
Muntoni F, Cau M, Ganau A, et al. Brief report: deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N Engl J Med 1993;329:921–925.
Li D, Tapscoft T, Gonzalez O, et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999;100:461–464.
Giordano C, D'Amati G . Cardiomyopathies due to defective energy metabolism: morphological and functional features. Pathologica 2005;97:361–368.
Hayashi T, Arimura T, Ueda K, et al. Identification and functional analysis of a caveolin-3 mutation associated with familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun 2004;313:178–184.
Kikuchi T, Oka N, Koga A, et al. Behavior of caveolae and caveolin-3 during the development of myocyte hypertrophy. J Cardiovasc Pharmacol 2005;45:204–210.
Koga A, Oka N, Kikuchi T, et al. Adenovirus-mediated overexpression of caveolin-3 inhibits rat cardiomyocyte hypertrophy. Hypertension 2003;42:213–219.
Ohsawa Y, Toko H, Katsura M, et al. Overexpression of P104L mutant caveolin-3 in mice develops hypertrophic cardiomyopathy with enhanced contractility in association with increased endothelial nitric oxide synthase activity. Hum Mol Genet 2004;13:151–157.
Woodman SE, Park DS, Cohen AW, et al. Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem 2002;277:38988–38997.
Cagliani R, Bresolin N, Prelle A, et al. A CAV3 microdeletion differentially affects skeletal muscle and myocardium. Neurology 2003;61:1513–1519.
Barbuti A, Terragni B, Brioschi C, et al. Localization of f-channels to caveolae mediates specific beta2-adrenergic receptor modulation of rate in sinoatrial myocytes. J Mol Cell Cardiol 2007;42:71–78.
Balijepalli RC, Foell JD, Hall DD, et al. Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci USA 2006;103:7500–7505.
Calaghan S, White E . Caveolae modulate excitation-contraction coupling and beta2-adrenergic signalling in adult rat ventricular myocytes. Cardiovasc Res 2006;69:816–824.
Reijneveld JC, Ginjaar IB, Frankhuizen WS, et al. CAV3 gene mutation analysis in patients with idiopathic hyper-CK-emia. Muscle Nerve 2006;34:656–658.
Hernandez-Deviez DJ, Martin S, Laval SH, et al. Aberrant dysferlin trafficking in cells lacking caveolin or expressing dystrophy mutants of caveolin-3. Hum Mol Genet 2006;15:129–142.
Matsuda C, Hayashi YK, Ogawa M, et al. The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum Mol Genet 2001;10:1761–1766.
Matsuda C, Aoki M, Hayashi YK, et al. Dysferlin is a surface membrane-associated protein that is absent in Miyoshi myopathy. Neurology 1999;53:1119–1122.
Fee DB, So YT, Barraza C, et al. Phenotypic variability associated with Arg26Gln mutation in caveolin3. Muscle Nerve 2004;30:375–378.
Fischer D, Schroers A, Blumcke I, et al. Consequences of a novel caveolin-3 mutation in a large German family. Ann Neurol 2003;53:233–241.
Parton RG . Caveolae and caveolins. Curr Opin Cell Biol 1996;8:542–548.
Schlegel A, Lisanti MP . A molecular dissection of caveolin-1 membrane attachment and oligomerization. Two separate regions of the caveolin-1 C-terminal domain mediate membrane binding and oligomer/oligomer interactions in vivo. J Biol Chem 2000;275:21605–21617.
Das K, Lewis RY, Scherer PE, et al. The membrane-spanning domains of caveolins-1 and -2 mediate the formation of caveolin hetero-oligomers. Implications for the assembly of caveolae membranes in vivo. J Biol Chem 1999;274:18721–18728.
Smythe GM, Rando TA . Altered caveolin-3 expression disrupts PI(3) kinase signaling leading to death of cultured muscle cells. Exp Cell Res 2006;312:2816–2825.
Acknowledgements
We thank Paolo Broda for expert technical assistance. This work was supported by Telethon-Italia, MURST and the Italian Ministry of Health (G. Gaslini Institute, Ricerca Finalizzata).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Traverso, M., Gazzerro, E., Assereto, S. et al. Caveolin-3 T78M and T78K missense mutations lead to different phenotypes in vivo and in vitro. Lab Invest 88, 275–283 (2008). https://doi.org/10.1038/labinvest.3700713
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700713
Keywords
This article is cited by
-
A novel 1-bp deletion variant in DAG1 in Japanese familial asymptomatic hyper-CK-emia
Human Genome Variation (2022)
-
T-tubule remodeling in human hypertrophic cardiomyopathy
Journal of Muscle Research and Cell Motility (2021)
-
Sudden cardiac death: focus on the genetics of channelopathies and cardiomyopathies
Journal of Biomedical Science (2017)
-
Identification and functional analysis of a new putative caveolin-3 variant found in a patient with sudden unexplained death
Journal of Biomedical Science (2014)
-
RNA Interference Improves Myopathic Phenotypes in Mice Over-expressing FSHD Region Gene 1 (FRG1)
Molecular Therapy (2011)
