
Abstract
X-linked adreno-leukodystrophy is a progressive, systemic peroxisomal disorder that primarily affects the adrenal cortex, as well as myelin and axons of the central nervous system. Marked phenotypic heterogeneity does not correlate with disease-causing mutations in ABCD1, which encodes a peroxisomal membrane protein that is a member of the ABC transmembrane transporter proteins. The precise physiological functions of ABCD1 and ABCD2, a closely related peroxisomal membrane half-transporter, are unknown. The abcd1 knockout mouse does not develop the inflammatory demyelination so typical and devastating in adreno-leukodystrophy, but it does display the same lamellae and lipid profiles in adrenocortical cells under the electron microscope as the human patients. The adrenocortical cells in the mouse also exhibit immunohistochemical evidence of oxidative stress at 12 weeks but no evidence of oxidative damage. To better understand the pathogenesis of this complex disease, we evaluate the adrenal lesion of the abcd1 knockout mouse as a function of normal aging, dietary or therapeutic manipulations, and abcd genotype. The loss of abcd2 causes oxidative stress in the adrenal at 12 weeks, as judged by increased immunoreactivity for the mitochondrial manganese superoxide dismutase, in both the inner cortex and medulla. The loss of abcd2 (n=20), but not abcd1 (n=27), results in the spontaneous and premature deposition of ceroid, a known end-product of oxidative damage, predominantly in adrenal medullary cells. These data indicate that the loss of abcd2 results in greater oxidative stress in murine adrenal cells than the loss of abcd1, providing a clue to its cellular function. We also find that the adrenocortical lesion of the abcd1 knockout mouse does not produce functional impairment at ten to nineteen months or overt hypocortisolism at any age, nor does it progress histologically; these and other data align this mouse model closer to human female heterozygotes than to male ALD or AMN hemizygotes.
Similar content being viewed by others


Introduction
X-linked adreno-leukodystrophy (XALD, ALD) is a progressive, systemic peroxisomal disorder that primarily affects the adrenal cortex, as well as myelin and axons of the central nervous system (CNS). The two major clinical phenotypes, often occurring in the same family, are: juvenile or cerebral ALD that shows profound inflammatory cerebral demyelination and an adult form termed adrenomyeloneuropathy (AMN) that is characterized by a distal-proximal (‘dying-back’) spinal axonopathy. Addisonian phenotypes, both isolated or more commonly in concert with ALD and AMN, also exist. The reason for this phenotypic divergence is unknown, but it does not correlate with mutations in the ABCD1 gene located at Xq28 (reviewed by Moser et al1). The identity of a putative modifier gene has remained elusive for over a decade. Both CNS phenotypes demonstrate the same primary non-inflammatory adrenocortical atrophy that appears to be due to apoptotic cell death associated with the accumulation of saturated unbranched very long chain (≥C22) fatty acids (VLCFA), predominantly esterified to cholesterol.2
ABCD1 encodes a peroxisomal membrane protein (ABCD1, ALDP) that is a member of the ATP Binding Cassette (ABC) transmembrane transporter proteins;3, 4 its physiological function is unknown, as is its pathogenetic role in ALD or AMN. ABCD2 (ALDR) at 12q1 codes for a closely related peroxisomal ABC integral membrane protein, ABCD2 or ALDRP. Its physiological function is also unknown; even though no known human disease is associated with this gene, overexpression of abcd2/ABCD2 can compensate for the loss of abcd1/ABCD1 in the mouse in vivo and in cultured human and mouse fibroblasts.5, 6 There is overlap of ABCD1 and ABCD2 expression in several tissues, but in the adrenal gland expression is reported to be a mirror-image: in the mouse adrenal gland, abcd1 is primarily expressed in cortex and abcd2 only in medulla.7 We have endeavored to clarify the function and pathologic role of abcd1 and abcd2 in the adrenal gland by examination of knockout (KO) mice lacking abcd1 and/or abcd2 generated by targeted homologous recombination.6, 8 Mice lacking abcd1 or abcd2 have failed to reproduce the inflammatory cerebral demyelination characteristic of human ALD.8, 9, 10 The major CNS lesion in the aged abcd1 KO (ALD) mouse, as well as in the aged abcd2 KO mouse and abcd1/abcd2 double KO mice, is spinal axonal degeneration that is reminiscent of AMN.6, 11, 12 However, the adrenocortical cells of our abcd1 KO mouse exhibit the same lamellar and lipid profiles, detectable with the electron microscope, as those seen in both human ALD and AMN (ALD/AMN).8
Recent studies of the abcd1 KO mouse in our laboratory have revealed some interesting and surprising findings.13, 14 (1) While elevated levels of VLCFA remain the biochemical signature of ALD/AMN, it is clear that this is not due to a deficiency in peroxisomal VLCFA degradation, as was previously believed. (2) The rate of peroxisomal β-oxidation of VLCFA in cultured fibroblasts is directly related to the rate of mitochondrial long-chain fatty acid β-oxidation. (3) The adrenocortical lesion does not appear to progress, as does that of the ALD/AMN patients, and the mice do not display overt hypocortisolism. (4) Mitochondrial abnormalities are present, which suggest a pathogenic role for oxidative stress. The latter prompted a comparative study of the abcd1 KO mouse and human ALD in which we found evidence of mild oxidative stress in the adrenal cortex of the abcd1 KO mouse, but no oxidative damage there or in its CNS at 12 weeks of age. This stands in striking contrast to human ALD where oxidative stress and mild oxidative damage were noted in the adrenal cortex, while profound oxidative damage was seen in the inflammatory cerebral demyelination.15 The reason for the apparent self-limiting nature of the abcd1 KO mouse adrenal lesion remained unclear; we theorized that its adrenocortical cells were able to adapt to the oxidative stress, at least temporarily (12 weeks), through the overexpression of the anti-oxidant mitochondrial manganese superoxide dismutase (formerly MnSOD, currently SOD2).
The normal, aged mouse adrenal gland, in contrast to its human counterpart, displays evidence of oxidative damage in the form of ‘lipogenic pigmentation’ or ‘brown degeneration’ of the inner adrenal cortex,16, 17 which can be detected through standard morphologic techniques. This pigmentation reflects the presence of an oxidative end-product, ceroid (reviewed by Yin18), so it is also referred to as ‘ceroid deposition’.17 Hence, this ceroid can serve as one line of evidence for oxidative damage in the murine adrenal.
In order to test the hypothesis that the abcd1 KO can contain the oxidative stress mentioned above, to determine if its adrenal lesion is truly static and remains subclinical, and to examine the functional relationship between abcd1 and abcd2 in a more simplified system (ie, the adrenal gland), we evaluated the abcd1 KO adrenal lesion as a function of normal aging, dietary or therapeutic manipulations, and abcd genotype. We also sought earlier evidence of oxidative stress/damage and a confirmation that ‘ceroid deposition’ is the result of oxidative damage through the immunohistochemical expression of heme oxygenase-1 (HO-1), malondialdehyde (MAL) and 4-hydroxynonenal (HNE). With these manipulations and techniques we find that: wild type (WT) 129Sv mice rarely develop ceroid at 30 months; the aged abcd1 KO (pure 129Sv background) mouse also rarely develops ceroid and then only minimally; and the abcd1 KO adrenal lesion neither progresses pathologically nor does it produce hypocortisolism. However, the abcd2 (also pure 129Sv) and abcd1/abcd2 double KO mice consistently and spontaneously display ceroid accumulations and other evidence of oxidative damage, even at younger ages. The significance of these differences is discussed.
Materials and methods
All animal housing and experimental manipulations were done in compliance with local institutional (JHMI) and NIH guidelines.
Establishment of Murine Knockouts
The generation of abcd1 and abcd2 KO was by targeted homologous recombination as previously described (8 and 6, respectively). Abcd2 knockout animals (129SvEvPas) were back crossed for 7–10 generations to the same 129SvEv mouse strain (Tac) used to generate the abcd1 mouse. Double abcd1 and abcd2 knockout animals were generated by cross breeding between the single knockout strains. All mice used in these studies were maintained on identical 129SvEv backgrounds and the abcd genotypes verified by molecular genotyping. The only exceptions were those used to determine the basal levels of ACTH and corticosterone of WT and abcd1 KO mice; these were of mixed 129SvEvTac/C57BL6 Tac genetic backgrounds.
Statistical Analysis
Statistical significance of comparison of biochemical parameters in Figures 6 and 7 was determined by calculation of P-values using Student's two-tailed t-test and SPSS statistics software (SPSS Inc., Chicago, IL, USA) for linear regression analysis.

Mouse adrenal function evaluation. Plasma (a) adrenocorticotropic hormone (ACTH, pg/ml) and (b) corticosterone (ng/ml) levels were measured before (basal) or one hour after stress (stressed) induced by vigorous cage shaking. (c) Kinetic studies of corticosterone secretion in response to stress stimulation. Plasma corticosterone levels from four WT and four abcd1 KO mice were measured at indicated time points following induced stress. While there were statistically significant differences between basal ACTH levels of WT and abcd1 KO mice, P<0.003, there were no other statistically significant differences between WT and abcd1 KO mice by either the Student's t-tailed t-test or linear regression analysis. n, number of samples measured. s.e. of measurements is indicated by error bars.

VLCFA levels in the adrenal of Lorenzo's oil treated abcd1 KO mice. Erucic acid (C22:1ω9; the active ingredient of Lorenzo's oil) and saturated VLCFA levels were measured. The y-axis represents the percentage of total fatty acids. n, number of samples measured. s.d. of measurements is indicated by error bars. For adrenal VLCFA levels of abcd1 KO mice before and after one month of Lorenzo's oil the P-values for C22:1ω9, C22:0, C24:0 and C26:0 were <0.005, 0.009, 0.445 and 0.034, respectively.
Morphologic Analyses
Adrenal glands were harvested from abcd KO, double KO and WT mice after perfusion with 25 ml ice-cold phosphate-buffered saline, pH 7.4, followed by 100 ml of a 4% glutaraldehyde and 4% paraformaldehyde mixture. Adrenal glands were either fixed and preserved in 4% glutaraldehyde, postfixed in osmium tetroxide, and embedded in epoxy resin or transferred to buffered Formalin (Richard-Allan Scientific, Kalamazoo, MI, USA) and processed routinely for paraffin embedding. The samples for electron microscopy were thin sectioned and stained with uranyl acetate-lead citrate.
The adrenal glands from WT mice (n=10; males; 9–30 months of age; average age=19.4 months), abcd1 KO (n=18; males; 4–28 months; average age=11.5 months), abcd2 KO (n=3; 21–23 months; males) and abcd1/abcd2 double KO (n=4; 19–20 months; males) were sectioned serially until the interface between the cortex and medulla was identified; usually, this consisted of a cross-section of the entire gland, but occasionally only a quadrant or half was available (Table 1A). These sections were stained with hematoxylin and eosin (H&E), as well as diastase-periodic acid-Schiff (D-PAS)-Luxol fast blue. The D-PAS is particularly effective for staining the lipogenic pigment. Selected adrenal cortices from each category (ie, abcd1 KO, WT, etc.) were also processed routinely for ultrastructural analysis. In order to eliminate the possibility that age or sex might be confounding variables, another group of untreated KO mice were processed for D-PAS staining: four abcd1 KO mice (three females, one male; 20–36 months), four abcd2 KO mice (all males and 22 months) and four abcd1/abcd2 KO mice (two males, two females and 19–23 months) (Table 1B). This untreated group was compared to the initial groups and to another group that had been treated with 4-phenylbutyrate (4-PBA) (10 g/l of drinking water for 2 months): two WT (both females and 22 months), five abcd1 KO (three females, two males; 26–31 months; average age=28.4 months), two abcd2 KO (female, male; 23 and 25 months, respectively), and three abcd1/abcd2 double KO (one female, two males; all 21 months) (Table 1C). 4-PBA has been shown to improve the saturated fatty acid defect in ALD and abcd1 KO fibroblasts.13, 14 Two ceroid-positive 21-month-old male abcd2 KO mice were stained with the Bodian silver protargol method and immunostained for synaptophysin (a medullary cell marker; rabbit monoclonal, 1:600; Neomarkers, Fremont, CA, USA), F4/80 (a mouse macrophage marker; rat monoclonal, 1:50 with heat retrieval; Caltag, Burlingame, CA), activated caspase-3, (polyclonal, 1:1000; R & D Systems, Minneapolis, MN, USA), heme oxygenase-1 (HO-1; polyclonal, 1:2000 with heat retrieval; Stressgen, Victoria, BC, Canada), HNE (polyclonal, 1:2750 with heat retrieval; Calbiochem, LaJolla, CA, USA) and MAL (polyclonal, 1:750 with heat retrieval; Stressgen, Victoria, BC, USA), along with one 24-month old WT and one 21-month-old male abcd1 KO.
In Situ Hybridization and SOD2-Immunohistochemical Methods in WT and Abcd2 KO Mice
Plasmids containing full-length cDNA encoding the murine abcd2 in pcDNA3 were linearized by cutting at a restriction site upstream of the initiator methionine codon (for antisense probes) or just downstream of the stop codon (for sense probes). Antisense RNA probes for abcd2 were synthesized using digoxigenin-labeled UTP (Roche Molecular Biochemicals, Indianapolis, IN, USA) and SP6 RNA polymerase (Ambion Inc., Austin, TX, USA). The corresponding digoxigenin-labeled sense probes were synthesized for use as controls using T7 RNA polymerase (Ambion Inc., Austin, TX, USA). The specificity of the abcd2 riboprobe used for in situ hybridization was established by Southern blot hybridization to total cDNA from abcd1 and abcd2 KO mouse fibroblasts. Both abcd1 and abcd2 are expressed in fibroblasts from WT mice. No hybridization was detected with cDNA for abcd2 KO mice showing that the abcd2 probe does not cross-hybridize with abcd1 cDNA.
Adrenal glands obtained from 3-month-old male WT mice were quick-frozen in liquid nitrogen. Eight micrometer sections were obtained using a Microm HM500M cryostat in RNase-free conditions, collected on Superfrost Plus slides (Fisher, Pittsburgh, PA, USA) that were pretreated with RNaseZAP (Sigma, St Louis, MO, USA). Slides were fixed for 30 min with 4% paraformaldehyde in phosphate-buffered saline, and acetylated for 10 min in 0.25% acetic anhydride in 0.1 M triethanolamine. After 8 h of prehybridization in 50% formamide containing 5 × Denhardt's solution (Sigma, St Louis, MO, USA), 5 × SSC, and 0.25 mg/ml yeast tRNA (Invitrogen, Carlsbad, CA, USA), slices were hybridized in the same buffer containing digoxigenin-labeled antisense or sense probes (200 ng/ml) for 16 h at 60°C. The slides were washed under high-stringency conditions (5 × SSC in 50% formamide for 5 min at 59°C, 2 × SSC for 1 min at 59°C, 0.2 × SSC for 30 min at 65°C, and 0.2% SSC for 5 min at room temperature). After a 1-h incubation in blocking reagent (Roche Molecular Biochemicals, Indianapolis, IN, USA), slices were incubated for 1.5 h with alkaline phosphatase-conjugated anti-digoxigenin antibody (Roche Molecular Biochemicals, Indianapolis, IN, USA). Color development with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate was carried out for 45 min without a hematoxylin counterstain. Sections were dehydrated and mounted with DPX-mounting solution (Fluka Biochemika, Milwaukee, WI, USA).
After fixation with 4% paraformaldehyde, frozen sections from 3-month-old male WT, abcd1 KO and abcd2 KO mice were incubated for 20 min with 0.6% H2O2 in methanol followed by 20 min with 5% normal goat serum. Sections were then incubated sequentially with Avidin D and biotin for 15 min each (Avidin/Biotin blocking kit, Vector Laboratories, Burlingame, CA, USA). Incubation with primary rabbit anti-SOD2 antibody (0.6 μg/ml, Stressgen, Victoria, BC, Canada; 1:250) was performed overnight at 4°C. Peroxidase-based detection was carried out using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA). After counterstaining with hematoxylin for 20 s, sections were dehydrated and mounted with DPX-mounting solution.
Adrenal Function Tests in Abcd1 KO Mice
All mice were of the 129SvEv genetic background, except for those used in the determination of basal levels of ACTH and corticosterone, who were of a mixed 129Sv and C57BL/6 background. The mice, matched for sex and age, were housed in groups. Those used for the basal and stress tests were 10–19 months of age. One week before testing of adrenal function the handler spent 30 min each day, between 0900 and 1000, to customize the mice to handling and to reduce stress. The evening before the experiment, the mice were moved to a separate room next to the laboratory. Individual mice were removed from the cage with minimal disturbance. The mice were cervically dislocated before or one hour after stress induced by vigorous cage shaking. The blood was immediately collected from sacrificed mice by heart puncture. For kinetic studies of corticosterone secretion, 3–6 month old mice were stressed, anesthetized immediately, and their blood collected by orbital bleeding at indicated time points. Plasma was prepared at room temperature by centrifugation of whole blood at 2000 × g for 5 min using EDTA as an anticoagulant. Adrenocorticotropic hormone (ACTH) levels were assayed using an ACTH 65T kit (Nichols Institute Diagnostics, San Clemente, CA, USA). Standards and mouse plasma were incubated with both a biotin coupled polyclonal antibody raised against the carboxy-terminal region of ACTH and an 125I labeled monoclonal antibody raised against the amino-terminal region of ACTH to form a soluble sandwich complex. The resulting sandwich complexes were then captured by avidin-coated beads. Plasma ACTH levels were determined by comparing bead bound radioactivity from plasma samples to that of the standards. Corticosterone levels were determined using an ImmuChem™ Double Antibody Corticosterone 125I RIA kit (MP Biochemicals LLC, Irvine, CA, USA). Briefly, equal amounts of 125I labeled corticosterone were used to compete for limited amounts of anti-corticosterone antiserum. The resulting immune complexes were precipitated, and their radioactivity measured. The amount of plasma corticosterone is inversely proportional to measured radioactivity.
Dietary Challenge of Abcd1 KO Mice and Treatment with Lorenzo's Oil and DHEAS
There is ample evidence that the signature free saturated fatty acids might be toxic to adrenocortical cells in ALD, and a threshold effect has been postulated (reviewed by Powers et al2). Peanut oil enriched mouse chow, at a final concentration of 20% (v/w), was prepared by adding peanut oil to regular mouse chow base, Purina Rodent Diet 5001 (Laboratory Rodent Diet, purchased from Bio Serve Inc., Frenchtown, NJ, USA). Four abcd1 KO mice (three males; one female; 9–13 months of age) had been treated with a high-saturated fat diet for 10–12 months. Lorenzo's oil (four parts Glycerol Trioleate oil and one part Glycerol Trierucate oil, SHS International Ltd, Liverpool UK) is known to rapidly reduce the level of saturated fatty acids in the blood of ALD/AMN patients (reviewed by Moser et al1). An enriched chow was prepared by soaking Purina Rodent Diet 5001 pellets in excess undiluted Lorenzo's oil for three days prior to feeding mice. It is estimated that 40–50 ml of Lorenzo's oil is either absorbed into or adsorbed onto every 100 g dry weight of Purina Rodent Diet 5001 mouse chow. Regular chow base with or without modifications was given to 6-month-old mice of matched age in control and test groups for 1–3 months. Androgens have been proposed to play a synergistic pathogenetic role in the onset of ALD/AMN (2). Two abcd1 KO mice were treated with the androgen dehydroepiandrosterone sulfate (DHEAS–1.5 mg/ml of drinking water for 6 months) starting 15 days after birth.
Biochemical Analyses of Abcd1 KO Mice
Plasma and tissue fatty acids from age-matched wild-type and abcd1 KO mice were analyzed. Plasma (50 μl) and one whole adrenal, homogenized in 50 μl water, from each animal were added separately to 13 × 100 mm2 glass screw capped tubes containing a solution of 1.0 ml 3:1 (v/v) methanol:methylene chloride solution (plus BHT 50 mg/l) plus 0.1 ml water. The samples were directly methanolyzed by adding 200 μl of fresh colorless acetyl chloride drop by drop, while vortexing. Then the tubes were capped and placed in an oven at 75°C. for 1 h. After cooling, 4.0 ml of 7% K2CO3 were added to stop the methanolysis reaction. The resulting total lipid fatty acid methyl esters were extracted twice with 2.0 ml of hexane, the hexane layers were combined, dried under nitrogen, dissolved in 100 μl hexane, transferred to injection vials and analyzed by capillary gas chromatography (GC).19
Fibroblast Studies
Fibroblasts from WT, abcd1 KO and abcd2 KO mice were cultured in modified essential media (MEM, Mediatech, Herndon, VA, USA) supplemented with 10% fetal calf serum (Mediatech, Herndon, VA, USA), penicillin (1 U/ml, Mediatech), and streptomycin (1 μg/ml, Mediatech) at 37°C in a 5% CO2 atmosphere. For hydrogen peroxide studies, fibroblasts were grown in the presence of 125 μM H2O2 for 96 h. To prepare fibroblasts for electron microscopy, cells were trypsinized, pelleted at 500 × g, resuspended in 1% glutaraldehyde in PBS pH 7.4 for 3 h, pelleted at 500 × g, and resuspended in 1.5 ml PBS pH 7.4. Osmium tetroxide (1%) was added for 20 min to the pellets. Prepared aqueous 3.0% agarose was heated to melting in a water bath. Melted agarose (2.0 ml) was used to resuspend cells in the conical tube after the last wash then quickly placed into a centrifuge to spin on high speed for 5 min. The agarose trapped cells were placed in the refrigerator to cool completely. The agarose/cells were removed from the tube whole and the blackened cell pellet tip was cutoff using a straight edged razor blade. The pellet was cut into 1.0 mm cubes/blocks and placed into vials and dehydrated in a graded series of ethanol to 100%, transferred into a 1:1 ratio of 100% ethanol/propylene oxide, then 100% propylene oxide (2 changes), and finally infiltrated with propylene oxide/EPON/Araldite epoxy resin, (1:1, 1:2) then two changes of 100% EPON/Araldite resin, the last one overnight. The cell blocks were placed into silicon molds containing fresh resin and polymerized for 2 days at 70°C. Semi-thin sections stained with toluidine blue and thin sections stained with uranyl acetate-lead citrate were examined for any group differences, particularly for the presence or absence of ceroid at the ultrastructural level.
Results
Adrenal ceroid (lipogenic pigmentation, brown degeneration) in aged mice is easily identifiable with the H&E stain as cells with swollen faint-yellow to beige cytoplasm, often appearing as multinucleated cells in which the nuclei may be compressed or distorted16 (Figure 1a). Thus, they resemble macrophages; but, in aged rodents, they usually arise from the innermost or juxtamedullary adrenocortical cells. They are more easily demonstrated by their intense and diffuse PAS- (Figure 1b) and acid-fast positivity. These cells are located primarily at the interface of the cortex and medulla, but may be found between the medullary cords17 (Figure 1c). Ultrastructurally, the ceroid in the cytoplasm of these cells consists of a heterogeneous mixture of membranous and granular materials,20, 21 distinctly different from classical lipofuscin and the lamellar-lipid profiles of human ALD/AMN or the abcd1 KO (Figure 2a).

(a) Lipogenic pigmentation. 21-month-old abcd2 KO mouse. Hematoxylin-eosin × 225. (b) Lipogenic pigmentation. Same 21-month-old abcd2 KO mouse. Luxol fast blue-D-PAS × 150. (c) Lipogenic pigmentation. Same 21-month-old abcd2 KO mouse. Cortex (blue, left) and medulla (pink, right) separated by intense PAS-positive deposits (++) located almost entirely with the medulla. Note the smaller PAS-positive inclusions in medullary cells (arrowheads). Luxol fast blue-PAS × 37.

(a) Lamellae and lipid profiles (arrows) in an adrenocortical cell. 13-month-old abcd1 KO mouse. Uranyl acetate-lead citrate. (b) Typical ceroid filling the cytoplasm of a probable cortical cell. 30-month-old WT mouse. Uranyl acetate-lead citrate. (c) Typical ceroid (arrow) within an inner cortical cell that is adjacent to another with cholesterol clefts (arrowheads). Six-month-old abcd1 KO treated with DHEAS for 6 months. Uranyl acetate-lead citrate. (d) Scant ceroid (+/−) within inner cortical cells (arrows). Twenty-eight-month-old abcd1 KO mouse treated with 4-PBA for 2 months. D-PAS × 150.
Ceroid in Abcd1, Abcd2, Abcd1/Abcd2 KO and WT Mice
None of the WT 129Sv mice, even at 30 months, display ceroid with H&E or D-PAS (Table 1A and C). However, a single cell at the cortico-medullary junction contains typical ceroid ultrastructurally in a 30-month-old male WT (Figure 2b).
Neither male nor female untreated abcd1 KO mice, even as old as 36 months (Table 1) spontaneously display ceroid at the light microscopic level, even with D-PAS. The four abcd1 KO mice treated with a high-saturated fat diet also fail to display ceroid with either the light or electron microscopes, but one of them contains numerous cholesterol clefts, most of which is within lysosomes, as previously reported.6 One of two 6-month old abcd1 KO mice treated with DHEAS contains a small amount of typical ceroid ultrastructurally (Figure 2c). One 28-month-old female abcd1 KO treated with 4-PBA demonstrates trace amounts of ceroid detectable only with D-PAS (Figure 2d).
The untreated abcd2 KO mice, all males, and most double KO mice, male and female, manifest abundant ceroid, even with only the H&E (see Figure 1a). The ceroid in one 21-month-old female double KO is detectable with D-PAS, but not with H&E. The ceroid is confirmed ultrastructurally in the abcd2 KO mouse, and it is associated with numerous cholesterol clefts (Figures 3a and b). The three ceroid-filled cells available for ultrastructural analysis have no identifiable mitochondria; but the mitochondria of both inner cortical and adjacent medullary cells are mildly swollen compared to those of the aged WT mouse.

(a) Ceroid-filled cell, possibly cortical, with compressed nucleus and cholesterol clefts (arrowhead). Same abcd2 KO mouse as in Figure 1. Uranyl acetate-lead citrate. (b) Higher magnification of another ceroid-filled cell with cholesterol clefts (arrowheads). No mitochondria are visible. Same abcd2 KO mouse. Uranyl acetate-lead citrate. (c) Small ceroid deposits (+) within inner cortical cells (arrows) adjacent to morphologically unremarkable cortical (top) and medullary (bottom) cells. Twenty-five-month-old abcd2 KO mouse treated with 4-PBA. D-PAS × 150. (d) Ceroid-filled (+) medullary cells (arrows) immediately surrounded by morphologically unremarkable medullary cells. Twenty-two-month-old abcd2 KO mouse. D-PAS × 75. (e) Early ceroid deposition admixed with a few cholesterol clefts (right side of cell) in a medullary cell identified by its neurosecretory granules (arrow) in a 22-month old abcd2 KO mouse. Uranyl acetate-lead citrate. (f) HNE-immunoreactive granules (arrows) in ceroid-filled cells. Another 21-month-old abcd2 KO mouse. Anti-HNE × 150.
The 4-PBA-treated groups exhibit similar trends, but the amount of ceroid is much attenuated (Table 1C). Both abcd2 KO mice (one male and one female, 25 and 23 months, respectively) and the female double KO contain suspicious inclusions with H&E that are confirmed as ceroid deposits with D-PAS (Figure 3c).
Cellular Site and Oxidative Nature of the Ceroid
The ceroid-containing cells are usually found within the medulla (Figure 3d) and adjacent to clusters of medullary cells with darker cytoplasm than the majority of the medullary cells. The dark medullary cytoplasm is argyrophilic (Bodian positive), illustrating their norepinephrine-containing nature.22 Their ultrastructural features (ie, displaying small, pleomorphic, extremely electron-dense granules) (Figure 3e) also support this contention.22 The cells displaying ceroid do not stain with antibodies to the adrenal medullary marker synaptophysin or mouse macrophages (F4/80). However, slender F4/80-immunoreactive processes often surround the ceroid-containing cells, and the latter are often in intimate contact with synaptophysin-immunoreactive medullary cells. Small D-PAS-positive ceroid deposits are frequently identified in both inner cortical and medullary cells (see Figures 1c, 3c and d). At the electron microscopic level the presence of many cholesterol clefts, rare cholesterol ester droplets, and many microvilli are further evidence for the cortical origin of some cells with ceroid, while neurosecretory granules identify a few others as medullary (see Figure 3e). Sparse ceroid-containing cells have a few activated caspase-3- and HO-1-immunoreactive cytoplasmic granules, but neither ‘normal’ cortical nor medullary cells are immunoreactive. The ceroid-containing cells are immunoreactive for both HNE and MAL (Figure 3f); again, the adjacent ‘normal’ cortical and medullary cells are not. There appears to be a mild, but probably insignificant, increase in HNE and MAL in the outer cortex of the abcd2 KO mice compared to the aged WT or abcd1 KO mice.
In Situ Hybridization and SOD2
The 3-month-old WT adrenal gland exhibits a strong to moderate signal for abcd2—mRNA in the adrenal medulla, but also a diffuse weak signal in the cortex (Figure 4a). Weak SOD2 immunoreactivity in the 3-month-old abcd2 KO is present in both the medulla and inner cortex (Figure 4b), while in the WT it is weak to essentially negative and restricted to the outer cortex (Figure 4c). The SOD2 immunoreactivities in the 3-month-old abcd1 KO was stronger and more widespread, but restricted to the cortex (data not shown)—as published previously.15

(a) abcd2—mRNA in 3-month-old WT mouse. Strong to moderate signal with the antisense probe in medulla, and a weaker signal in surrounding cortex (top left and bottom). There is no hematoxylin counterstain. The sense probe was entirely negative (data not shown) × 100. (b) Weak signal in both medulla (arrows) and inner cortex (double arrow) of 3-month-old abcd2 KO. Anti-SOD2 X 100. Hematoxylin counterstain. (c) Extremely weak signal in outer cortex (arrow) of 3-month-old WT mouse. Anti-SOD2 × 100. Hematoxylin counterstain.
Fibroblast Studies
No obvious cytopathic changes were observed in WT, abcd1 KO or abcd2 KO fibroblasts. The fibroblasts of the abcd2 KO and WT grew faster than those of the abcd1 KO. Ultrastructurally, small deposits of typical ceroid were present rarely in both abcd1 KO and abcd2 KO fibroblasts. In the abcd2 KO fibroblasts a possible transition between the ceroid (Figure 5a) and abnormal mitochondria (Figure 5b) could be detected. Treatment with H2O2 did not result in any significant abnormalities or differences from the untreated cultures.

(a) An abcd2 KO fibroblast displaying several deposits of ceroid (arrows). Uranyl acetate-lead citrate. (b) An enlargement of the boxed area in Figure 6a showing markedly abnormal mitochondria, some still displaying the characteristic double unit membrane (arrows). Uranyl acetate-lead citrate.
Adrenal Functional Analyses in Abcd1 KO Mice
Evaluation of adrenal function in mice was evaluated by measurement of ACTH and corticosterone levels before and 1 h after stress. As shown in Figure 6a, abcd1 KO mice had an apparent 1.8-fold higher basal level than normal mice, 416.4±122.3 and 233.9±65.0 pg/ml, respectively. These differences are not statistically significant, P=0.22. After stress induced by vigorous cage shaking, the ACTH levels were significantly elevated in both abcd1 KO and normal mice (665.6±234.6 and 787.7±119.9 pg/ml, respectively; P=0.35 for KO and 0.004 for normal mice). There is no statistically significant difference between abcd1 KO and normal mice. Corticosterone levels increased in response to ACTH levels. Figure 6b shows that the basal corticosterone levels were equivalent in abcd1 KO and normal mice (54.3±6.0 and 46.1±8.5 ng/ml, respectively; P>0.2 for each). In response to ACTH stimulation following stress, the corticosterone levels increased approximately sixfold in both abcd1 KO and normal mice (366.8±77.4 and 293±29.4 ng/ml, respectively; P<0.003 for each). While there are significant differences between basal and stressed levels of both ACTH and corticosterone, there are no statistically significant differences between abcd1 KO and normal mice. A test of the statistical significance of the coefficients of variation using linear regression, which takes into account interactions between disease and stress status, give P-values of 0.829 for ACTH and 0.295 for corticosterone as a function of disease; 0.011 for ACTH and <0.005 for corticosterone as a function of stress; and 0.285 for ACTH disease/stress interaction and 0.397 for corticosterone disease/stress interaction. Thus, the presence or absence of abcd1 had no apparent effect on either the basal or stressed induced levels of ACTH or corticosterone.
As the data in Figure 6a and b were steady-state levels, kinetic analyses of corticosterone secretion in response to stress were measured, Figure 6c. Stress induced secretion of corticosterone into plasma was observed in both abcd1 KO and normal mice that persisted for up to 2.5 h. Whereas the secretion increase may be slightly slower in abcd1 KO mice for the first hour after stress, similar plateaus are achieved and there are no statistically significant differences between the mice at any of the time points, P>0.3 (P-values ranged from >0.31 to 0.88). Therefore, the rate of secretion of corticosterone in response to stress is not influenced by abcd1.
VLCFA Analysis of Mouse Chow
To estimate the VLCFA intake of abcd1 KO mice, the VLCFA content of the mouse chow regularly consumed by abcd1 KO mice in the laboratory was determined. The abundance of saturated VLCFAs (0.6%; C22:0=0.27%, C24:0=0.20%, C26:0=0.05% of total fatty acids) in mouse chow is relatively low and the amount of mono-unsaturated fatty acids (30.2%; C18:1ω9=26.15%, C20:1ω9=0.46%) is high. To conduct a dietary stress experiment, the regular 4.5% lipids in mouse chow was increased to 20% by addition of peanut oil that contains a higher amount of saturated VLCFAs (4.7%; C22:0=3.1%, C24:0=1.7%, C26:0=0.27%, ie about 1.6 mg/g of chow). This diet is estimated to increase dietary saturated VLCFAs approximately 40-fold.
VLCFA Analyses in Dietary Stressed and Lorenzo's Oil Treated Abcd1 KO Mice
VLCFA levels in plasma and adrenal glands of normal and abcd1 KO mice were measured after dietary stress with chow containing high levels of VLFCA (peanut oil). Plasma VLCFA levels after 11 months on the diet did not vary as a function of the presence or absence of abcd1 and did not differ from untreated controls (data not shown).
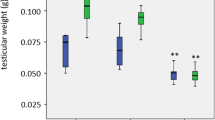
To investigate the effect of Lorenzo's oil (LO) on adrenal VLCFA levels, LO was mixed with mouse chow and fed to abcd1 KO mice for up to 3 months. Results are presented in Figure 7. There was a more than 30-fold increase in erucic acid, (C22:1ω9), the active ingredient in LO, observed at 1 and 3 months. The increase in erucic acid was associated with decreases in the measured saturated VLCFA, C22:0, C24:0 and C26:0. All decreases, except C24:0, were significant with P<0.05.
Discussion
Our present study confirms both the presence and the paucity of lipogenic pigmentation in the adrenals of aged mice.17 The loss of abcd1 does not significantly alter this course, even with dietary and therapeutic manipulations. The loss of abcd2, however, results in an acceleration of ceroid deposition in the adrenal gland. The additional loss of abcd1 in the double KO mice does not seem to augment this phenomenon.
Ceroid is a pathologic end-product of oxidative damage (reviewed by Yin18 and Porta23). It is enhanced by a dietary deficiency in antioxidants, such as vitamin E,23 or when mitochondrial damage is induced with propylthiouracil in the rodent24 and with o, p′ dichlorodiphenyldichloroethane (DDD) in the dog.25 In both the propylthiouracil-induced brown degeneration in the mouse adrenal and the o, p′ DDD intoxication of the dog adrenal, early abnormalities in mitochondrial ultrastructure (ie, swelling and loss of cristae) and ultimately their loss have been reported. Likewise, in the ceroid lipopigment deposition in Purkinje cells due to vitamin E deficiency, mitochondrial abnormalities are seen and believed to result in excessive free radical production.26 In the abcd2 KO mice, mitochondrial swelling in both cortical and medullary cells was observed, and the cells filled with ceroid displayed no mitochondria. Conversely, the accelerated course of ceroid deposition when abcd2 is absent appears to be slowed somewhat by 4-PBA treatment, which previously was shown to augment mitochondrial activity in the abcd1 KO.13, 14 Thus, it is reasonable to conclude that damaged mitochondria are a major contributor to these ceroids. The abcd2 KO fibroblast cultures also support this conclusion (see Figure 6b). Our study provides further evidence for oxidative damage being operative in the formation of this ceroid by the presence of HNE and MAL, toxic aldehydes derived from the oxidation of unsaturated lipids.27
Previous studies have shown that ceroid accumulates in aged rodent (mouse, rat) adrenal cells.16, 17, 20, 21 For the untreated C57BL/6, none is detected until 24 months of age, at which time approximately 10% of females and 5% of males demonstrate some.17 Treatment of this same strain with the estrogens, diethylstilbestrol and ethinyl estradiol, markedly increases the incidence of ceroid deposition in a dose-dependent manner with a weakened, but persistent, female preponderance.17 In the normal aged mouse ceroid is deposited in cells at the interface of the cortex and medulla. There is convincing evidence that it is deposited primarily in cortical,20, 21 but also in medullary, cells.21 Published data on ceroid deposition in the 129Sv adrenal could not be found. Based on our experiments this strain appears to be more resistant than C57 BL/6. Some differences exist between 129 substrains,28 but those between Tac and Pas are relatively insignificant. Moreover, the number of back crosses that were performed would ensure a uniform genetic background between groups. It is noteworthy that the same pattern has been observed in mice derived from breeding abcd2 KO mice onto C57BL/6 and 129SvEvPas mixed genetic background, estimated to be 50%, 75% and 100% C57 BL/6, but not when abcd1 is deleted. In this setting, ceroid inclusions are already detectable at 6 months (Pujol A, unpublished observations).
In the abcd2 or abcd1/2 double KO mice, ceroid is also found in both cell types, but probably more in medullary cells than in the normal aged rodent. Abcd2-mRNA is predominantly expressed by medullary, but also by cortical, cells (see Figure 4b). The only published expression data7 state that no abcd2 (ALDRP) is present in cortex, but its Figure 3b appears to show a weak cortical signal. Oxidative stress in both the cortex and medulla can be detected by the overexpression of SOD2 in the abcd2 KO at 3 months of age, while oxidative damage in the form of ceroid is prominent at least as early as 19–21 months in the abcd2 or abcd1/2 double KO mice. The distribution of ceroid in these KO mice most closely approximates that of norepinephrine-containing cells (22, its Figure 1), and the rare ceroid-containing cell identified ultrastructurally as medullary had neurosecretory granules consistent with norepinephrine (22, our Figure 3e). The norepinephrine-metabolite, DOPEGAL, generates free radicals;29 aging cells tend to produce more reactive species than their younger counterparts. Abcd2 might be needed to minimize or neutralize such toxic reactive species by importing some detoxifying element or cofactor into the peroxisomes of adrenal cells. The lack of any demonstrable change after H2O2 suggests that the loss of abcd2 does not impact directly on catalase activity. In the absence of abcd2, oxidation of the unsaturated fatty acids within cell membranes (eg, arachidonic),27 including those of mitochondria, appears to occur. Irrespective of the precise molecular details, our data demonstrate that the loss of abcd2 accelerates the deposition of ceroid in the 129Sv murine adrenal gland, suggesting that abcd2 (ALDRP), at least in the adrenal, may be more necessary for the control of oxidative stress than abcd1 (ALDP). The presence of numerous cholesterol clefts within the ceroid raises the additional possibility that abcd2, and to lesser extent abcd1, might play a role in intracellular cholesterol trafficking. Whether the ceroid deposition seen in the absence of abcd2, but not of abcd1, might be related to the excess of C22:0 reported only in the former6 remains unclear.
What then is the relevance of the abcd1 KO mouse as a model of the human disease: ALD or AMN? We have neither evidence that the loss of abcd1 (ALDP) in the mouse leads to significant adrenal atrophy, nor that these mice develop clinical signs of hypocortisolism, which commonly occur in ALD/AMN patients (reviewed by Moser et al1), in spite of a two- to four-fold increase in C26:0.6, 8 The possible onset of preclinical primary adrenocortical insufficiency suspected at 3–6 months (Figure 6c) is not confirmed at 10–19 months (Figures 6a and b). Our attempt to increase VLCFA in the KO adrenal cortex by dietary stress and possibly trigger a threshold response was unsuccessful. The widespread distribution of ballooned striated adrenocortical cells, so typical of ALD/AMN,30 also does not occur in our abcd1 KO mouse (nor the abcd1/2 double KO). Scattered striated cells, occasionally ballooned, are seen at the light microscopic level, and the identical lamellar-lipid profiles as in the human disease (see Figure 2a) are observed ultrastructurally as early as 3 months of age.5 Based on the available data (i.e., clinical, functional and pathologic) from the nervous system and adrenal,8, 9, 10, 11, 13, 14, 15 we believe that the abcd1 KO mouse best recapitulates the myelopathic ALD female heterozygote, not the male ALD/AMN hemizygotes.1, 31
References
Moser HW, Smith KD, Watkins PA, et al. X-linked adrenoleukodystrophy. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill: New York, NY, 2001, pp 3257–3301.
Powers JM, Schaumburg HH, Johnson AB, et al. A correlative study of the adrenal cortex in adreno-leukodystrophy—Evidence for a fatal intoxication with very long chain saturated fatty acids. Invest Cell Pathol 1980;3:353–376.
Mosser J, Douar AM, Sarde CO, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 1993;361:726–730.
Mosser J, Lutz Y, Stoeckel ME, et al. The gene responsible for adrenoleukodystrophy encodes a peroxisomal membrane protein. Hum Mol Genet 1994;3:265–271.
Netik A, Forss-Petter S, Holzinger A, et al. Adrenoleukodystrophy-related protein can compensate functionally for adrenoleukodystrophy protein deficiency (X-ALD): implications for therapy. Hum Mol Genet 1999;8:907–913.
Pujol A, Ferrer I, Camps C, et al. Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Hum Mol Genet 2004;13:2997–3006.
Troffer-Charlier N, Doerflinger N, Metzger E, et al. Mirror expression of adrenoleukodystrophy and adrenoleukodystrophy related genes in mouse tissues and human cell lines. Eur J Cell Biol 1998;75:254–264.
Lu J-F, Lawler AM, Watkins PA, et al. A mouse model for X-linked adrenoleukodystrophy. Proc Natl Acad Sci USA 1997;94:9366–9371.
Forss-Petter S, Werner H, Berger J, et al. Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J Neurosci Res 1997;50:829–843.
Kobayashi T, Shinnoh N, Kondo A, et al. Adrenoleukodystrophy protein-deficient mice represent abnormality of very long chain fatty acid metabolism. Biochem Biophys Res Commun 1997;232:631–636.
Pujol A, Hindelang C, Callizot N, et al. Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet 2002;11:499–505.
Ferrer I, Kapfhammer JP, Hindelang C, et al. Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late-onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum Mol Genet 2005;14:3565–3577.
McGuinness MC, Lu J-F, Zhang H-P, et al. Role of ALDP (ABCD1) and mitochondria in X-linked adrenoleukodystrophy. Mol Cell Biol 2003;23:744–753.
Heinzer AK, McGuinness MC, Lu J-F, et al. Mouse models and genetic modifiers in X-linked adrenoleukodystrophy. Adv Exp Med Biol 2003;544:75–93.
Powers JM, Pei Z, Heinzer AK, et al. Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol 2005;64:1067–1079.
Dunn TB . Normal and pathologic anatomy of the adrenal gland of the mouse, including neoplasms. J Natl Cancer Inst 1970;44:1323–1389.
Frith CH . Lipogenic pigmentation, adrenal cortex, mouse. In: Jones TC, Capen CC, Mohr U (eds). Endocrine Systems. Springer-Verlag: Berlin Heidelberg, Germany, 1996, pp 458–462.
Yin D . Biochemical basis of lipofuscin, ceroid, and age pigment-like fluorophores. Free Radic Biol Med 1996;21:871–888.
Moser AB, Jones DS, Raymond GV, et al. Plasma and red blood cell fatty acids in peroxisomal disorders. Neurochem Res 1999;24:187–197.
Samorajski T, Ordy JM . The histochemistry and ultrastructure of lipid pigment in the adrenal glands of aging mice. J Gerontol 1967;22:253–267.
Schardein JL, Patton GR, Lucas JA . The microscopy of ‘brown degeneration’ in the adrenal gland of the mouse. Anat Rec 1967;159:291–310.
Tramezzani JH, Chiocchio S, Wassermann GF . A technique for light and electron microscopic identification of adrenalin- and noradrenalin-storing cells. J Histochem Cytochem 1964;12:890–899.
Porta EA . Dietary factors in lipofuscinogenesis and ceroidogenesis. Arch Gerontol Geriatr 2002;34:319–327.
Moore NA, Callas G . Observations on the fine structure of propylthiouracil-induced ‘brown degeneration’ in the zona reticularis of mouse adrenal cortex. Anat Rec 1975;183:293–302.
Powers JM, Hennigar GR, Grooms G, et al. Adrenal cortical degeneration and regeneration following administration of DDD. Am J Pathol 1974;75:181–194.
Bertoni-Freddari C, Fattoretti P, Casoli T, et al. Morphometric investigations of the mitochondrial damage in ceroid lipopigment accumulation due to vitamin E deficiency. Arch Gerontol Geriatr 2002;34:269–274.
Esterbauer H, Schaur RJ, Zollner H . Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991;11:81–128.
Simpson EM, Linder CC, Sargent EE, et al. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet 1997;16:19–27.
Burke WJ, Kristal BS, Yu BP, et al. Norepinephrine transmitter metabolite generates free radicals and activates mitochondrial permeability transition: a mechanism of DOPEGAL-induced apoptosis. Brain Res 1998;787:328–332.
Powers JM, Schaumburg HH . Adreno-leukodystrophy (sex-linked Schilder's disease). Am J Pathol 1974;76:481–500.
Powers JM, Moser HW, Moser AB, et al. Pathologic findings in adrenoleukodystrophy heterozygotes. Arch Pathol Lab Med 1987;111:151–153.
Acknowledgements
The authors express their appreciation to Frances Vito for histologic and immunohistochemical support, Jenny Smith for graphic expertise and Tina Blazey for her usual outstanding secretarial assistance. They also recognize the support of Dr Elizabeth Metzger, Mouse Facility manager of the IGBMC, Strassbourg, for the care of the abcd2 KO mice and Dr Chung Li of the Department of Public Health, Fu Jen Catholic University, for helpful discussions regarding linear regression analysis.
Grant Support: This work was supported by Public Health Service Grants HD 10981, HD 24061 and NS 37355 from the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lu, JF., Barron-Casella, E., Deering, R. et al. The role of peroxisomal ABC transporters in the mouse adrenal gland: the loss of Abcd2 (ALDR), Not Abcd1 (ALD), causes oxidative damage. Lab Invest 87, 261–272 (2007). https://doi.org/10.1038/labinvest.3700512
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700512
