
Abstract
A growing number of tumors are characterized by simple genetic changes that activate important biochemical pathways, which are involved in their pathogenesis. These findings have led to the concept of targeted small molecule inhibitor treatment. The prototype for this type of therapy has been treatment of chronic myelogenous leukemia with imatinib mesylate (Gleevec), which targets BCR-ABL kinase. More recently, imatinib has been used to inhibit KIT in gastrointestinal (GI) stromal tumor, a mesenchymal tumor that arises in the GI tract. Furthermore, it has been possible to target EGFR in non-small-cell lung cancer with gefitinib and erlotinib. While initial results have been encouraging, resistance to small molecule kinase inhibitors is a substantial drawback. This paper focuses on what is known about mechanisms of resistance in the treatment of solid tumors by small molecule kinase inhibitors.
Similar content being viewed by others

Main
Only recently, have we fully appreciated that a subset of cancers can be largely dependent on a single oncogenic pathway. This has led to the oncogene addiction hypothesis.1 In a formal test of this hypothesis, Druker et al2 proposed that inhibition of ABL-kinase might inhibit the growth of chronic myelogenous leukemia (CML), which is characterized by the BCR-ABL fusion oncoprotein in more than 90% of the cases. This strategy has been very successful as imatinib mesylate (STI-571, Gleevec/Glivec, Novartis Pharmaceuticals, Basel, Switzerland), a small molecule inhibitor of ABL kinase, has become the treatment of choice for BCR-ABL positive CML.3 These results have been extended to solid tumors by targeting KIT in gastrointestinal stromal tumors (GIST),4 and epidermal growth factor receptor (EGFR) in non-small-cell lung cancer (NSCLC).5 Unfortunately, while these therapies have been successful initially, it has become clear that monotherapies designed to inhibit oncogenic kinases are unable to completely eradicate these tumors due to the development of resistance. This review summarizes the current state of what is known about mechanisms of resistance to small molecule inhibitors of oncogenic kinases in solid tumors.
Background
GIST is characterized by the presence of oncogenic KIT or platelet-derived growth factor receptor alpha (PDGFRA) mutations.6 KIT and PDGFRA are receptor tyrosine kinases with important roles in development, proliferation and cell death of many normal cell types, including interstitial cells of Cajal, the putative precursors to GIST. Mutations cluster within several exons of KIT (exons 9, 11, 13, and 17) and PDGFRA (exons 12, 14, and 18), preserve the open reading frame and result in ligand independent, constitutive activation of the kinase. These mutations are found within sporadic GISTs and also in rare familial GIST patients.6 Furthermore, mice harboring corresponding mutations in their germ line develop GISTs.8, 9 Imatinib mesylate, also potently inhibits KIT and PDGFRA, which led to its rapid FDA approval for treatment of metastatic GISTs after having shown considerable promise in clinical trials.
Approximately, 10% of NSCLCs are characterized by mutations in EGFR (epidermal growth factor receptor), a receptor tyrosine kinase with important roles in normal and malignant cell growth.5 Within this group, non-smoking women of East Asian background with adenocarcinoma are overrepresented.10 Mutations cluster within the ATP binding pocket in exons 18–21 of EGFR. As with GIST, these mutations preserve the open reading frame and encode constitutively active kinases. NSCLCs harboring these mutations are sensitive to the EGFR inhibitors gefitinib (Iressa, AstraZeneca, Wilmington, DE, USA) and erlotinib (Tarceva, OSI Pharmaceuticals, Melville, NY, USA).11
Primary resistance
Primary resistance refers to patients who either do not achieve stable disease or who progress within 6 months after an initial clinical response. In GIST, primary resistance to imatinib is seen predominantly in tumors with a KIT exon 9 mutation, most PDGFRA mutations, or no detectable kinase mutation (KIT and PDGFRA wild-type tumors).12, 13 At this point, the mechanism for primary resistance due to exon 9 mutations is unknown. However, there is some evidence that increasing the dose of imatinib might be able to overcome this resistance.14 In contrast to KIT mutations, primary PDGFRA mutations often affect the kinase domain, presumably leading to conformational changes that confer primary imatinib resistance.15
Similar to what is seen in GIST without KIT or PDGFRA mutations, NSCLC without EGFR mutations are also refractory to treatment with EGFR inhibitors.11 K-ras mutations are found in approximately 30% of NSCLC and these tumors do not contain EGFR mutations.16 K-ras, is known to be a downstream signaling molecule in the EGFR pathway. Tumors harboring K-ras mutations appear to be insensitive to therapy with EGFR inhibitors.16
Secondary resistance
Secondary resistance to small molecule inhibition typically occurs after prolonged treatment, and several molecular mechanisms have been suggested to contribute to the resistance phenotype. However, only a few have been experimentally proven. The most common mechanism leads to structural alterations in the kinase domain of the originally affected kinase. This results in the inability of the inhibitor to bind to and inhibit the catalytic activity of the kinase. The primary kinase mutation is always maintained.
Secondary mutations in the kinase domain of KIT or EGFR arise in approximately 50% of patients with GIST or NSCLC that have acquired clinical resistance to imatinib or gefitinib/erlotinib therapy, respectively.12, 17, 18, 19, 20, 21, 22, 23, 24
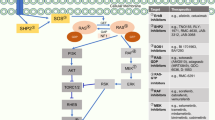
Different regions of the kinase domain are affected by secondary mutations, and much of what has been described in imatinib-resistant CML also applies to GIST and NSCLC. A common mutation affects the so-called gatekeeper residue of the BCR-ABL fusion kinase, which is conserved in KIT and EGFR and is also found to be mutated in GIST and NSCLC, respectively (see Figure 1).25 The gatekeeper residue consists of a threonine outside the actual catalytic core of the kinase. It is important for high-affinity binding to the inhibitor with which it forms a critical hydrogen bond. Most mutations at this site are predicted to abolish the ability to form this hydrogen bond. Additionally, a larger amino-acid side chain is introduced, leading to sterical hindrance.25 Threonine to methionine mutations of the gatekeeper residue of EGFR (T790M) were seen in all but one NSCLC that developed resistance to gefitinib to date.22, 23, 24 In GIST, the gatekeeper residue is mutated in about 20% of cases with a known resistance mutation and typically leads to substitutions of the original threonine by isoleucine (T670I) or phenylalanine (T670F).12, 17, 20, 21 This codon is located in a region (exon 14) where no primary KIT mutations have been found so far.

(a) Diagrammatic representation of oncogenically activated receptor tyrosine kinase, (b) inhibition of oncogenically activated kinase by small molecule inhibitor (KI), (c) resistance to small molecule inhibition secondary to a mutation that disrupts a critical hydrogen bond between the kinase inhibitor and the receptor tyrosine kinase and has a bulky side chain that interferes with binding (‘gatekeeper mutation’).
The most common secondary mutations in GISTs are found in KIT exons 13 and 17, both with a frequency of approximately 40%.12, 18, 19, 20, 21 These regions correspond to the ATP-binding and phosphotransferase domains, also referred to as p-loop and activation loop. Similar mutations have been detected in CML patients, but are less common. These mutations presumably destabilize the closed (or inactive) conformation of the kinase and remove hydrophobic contacts.25, 26 As imatinib preferentially binds the inactive conformation of KIT, this is a potential ‘Achilles’ heel’ that the cancers have exploited to develop resistance.25 While the p-loop mutation in GIST is always a valine to alanine substitution at residue 654 (V654A), which is never seen in primary GIST, exon 17 mutations are variable. They cluster around codon 820 (816–823) and have been found in primary GISTs. Of note, a mutation involving KIT codon 816 (D816V) is also known to be associated with mast cell neoplasms and is resistant to imatinib inhibition.27
No secondary PDGFRA mutations have been described to date in GISTs that present with a primary PDGFRA mutation. The reason for this might lie in the fact that these mutations are less common and that PDGFRA mutations more frequently show primary resistance to imatinib.
Another mechanism that can also contribute to the development of resistance is generically known as ‘kinase switch’.28 This mechanism, in which tumor cells activate a different kinase than the primary, targeted kinase, has been seen in GIST. Cases have been described that presented with a primary mutation in KIT exon 11 and acquired a secondary mutation in the kinase domain of PDGFRA,12, 28 a mutation that is also found in primary imatinib-resistant GISTs.7, 15 It is therefore likely that other, yet to be determined kinases including downstream effectors could also be activated in a similar manner in tumors targeted by kinase inhibitors.
Gene amplifications of BCR-ABL have been shown to contribute to secondary resistance in CML25 and were recently described for KIT and PDGFRA in GISTs.12, 28 These presumably lead to higher expression levels, resulting in the need for higher doses of inhibitor to sufficiently inhibit the target. The situation is not as clear in NSCLC where EGFR gene amplifications can already be detected in tumors before onset of treatment.29
EGFR receptor internalization, however, has been experimentally shown to play a role in secondary resistance in NSCLC.24 Drug-resistant cells showed altered receptor trafficking and demonstrated continued dependence on EGFR signaling without containing secondary EGFR mutations.
Additional mechanisms that can at least contribute to the resistant phenotype have been proposed, but most of them still need to be better defined. These include extracellular sequestration of the drug by plasma proteins as well as an enhanced active efflux by transmembrane pump proteins such as multidrug resistance (MDR)/p-glycoprotein.25, 30
Strategies to overcome resistance
A number of strategies to overcome secondary drug resistance are currently being tested (summarized in Table 1). They can be divided into a search for compounds that target oncogenically activated kinases or drugs that inhibit downstream effectors. Interference with the activity of the oncogenic kinase can be accomplished through (i) more specific kinase inhibitors, (ii) multitargeted kinase inhibitors, (iii) compounds that act differently from classical ATP-competitive binding, and (iv) compounds that reduce kinase expression or protein levels.
New kinase inhibitors have been developed that have an increased specificity for their target and the ability to inhibit kinases with resistance mutations. The rationale to design these more effective inhibitors is clear, especially in the case of imatinib. Compounds that bind to the oncogenic kinase in its active state (in contrast to imatinib) would be envisioned to be more efficient in target inhibition. New ABL-specific inhibitors have been developed that are 30- to 300-times more potent than imatinib and that bind to ABL in its active conformation.25, 31 Some of these compounds also inhibit KIT and PDGFRA and have entered clinical trials for GIST.32, 33
A number of compounds are being developed that intentionally inhibit a broader spectrum of kinases (multitargeted inhibitors). The underlying principle is to reduce the formation of resistant cell clones by inhibiting two unrelated oncogenic mechanisms. For example, simultaneously targeting the desired oncogenic kinase plus vascular endothelial growth factor (VEGF) and PDGF receptors to inhibit the formation of new blood vessels is thought to increase the antitumor effects. Sunitinib malate (Sutent, SU11248, Pfizer, NY, USA), a multitargeted inhibitor that effectively blocks KIT, PDGF and VEGF receptors, RET and FLT3, was recently FDA-approved for the treatment of imatinib-resistant GIST (and metastatic renal cell carcinoma). It has also entered a phase II clinical trial for patients with advanced NSCLC.31, 34
Several new compounds aim at inhibiting kinases by means other than competitively binding to the ATP-binding region. EGFR inhibitors that irreversibly bind to the receptor seem to be especially useful in NSCLC. A few compounds of this class have been shown to successfully inhibit the T790M resistance mutation, and some are in clinical trials.22, 24, 35 A new group of allosteric inhibitors that impede the kinase by binding distantly to the active site have recently gained interest.36 They putatively bind to the myristoyl pocket, exhibit exceptional target specificity and have synergistic antiproliferative effects when combined with imatinib.
A last group of compounds, characterized by heat shock protein (HSP) 90 inhibitors, does not target the phosphorylation mechanism of oncogenic kinases. HSPs are molecular chaperones that guide the normal folding, intracellular disposition and proteolytic turnover of many proteins.37 Oncogenic mutations often lead to conformationally unstable proteins that need excessive amounts of chaperones to be stabilized. Consequently, inhibition of these chaperones should lead to increased proteasomal degradation of these mutant proteins.37 HSP90 inhibitors have shown promising results in preclinical studies with GIST and NSCLC, and clinical trials have been initiated.38, 39
A growing group of compounds that might help to overcome secondary resistance aims at the inhibition of signaling molecules downstream of oncogenically activated kinases. Important effectors of most tyrosine kinases are the PI3K/AKT/mTOR and RAS/RAF/MAPK pathways, respectively.31, 40 Examples in this category are inhibitors that target mTOR and are currently being tested in clinical trials for GIST.41 Moreover, inhibitors of PI3K and RAS are currently under investigation in gefitinib-resistant NSCLC.42, 43
Finally, there are several possibilities of combination therapies that have in part found their way into clinical trials. These include regimens of the original compound plus conventional radio- or chemotherapy as well as treatments with the original drug plus the addition of one of the new compounds described above.
Summary
We have now entered the era of targeted therapy with small molecule inhibition of solid tumors. While primary resistance and secondary resistance will continue to be a problem, what we have learned so far has paved the way for further advances in this exciting area. The future looks bright for the treatment of solid tumors.
References
Weinstein IB . Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science 2002;297:63–64.
Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996;2:561–566.
Talpaz M, Silver RT, Druker BJ, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood 2002;99:1928–1937.
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–480.
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–2139.
Rubin BP . Gastrointestinal stromal tumours: an update. Histopathology 2006;48:83–96.
Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–710.
Rubin BP, Antonescu CR, Scott-Browne JP, et al. A knock-in mouse model of gastrointestinal stromal tumor harboring kit K641E. Cancer Res 2005;65:6631–6639.
Sommer G, Agosti V, Ehlers I, et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci USA 2003;100:6706–6711.
Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497–1500.
Janne PA, Engelman JA, Johnson BE . Epidermal growth factor receptor mutations in non-small-cell lung cancer: implications for treatment and tumor biology. J Clin Oncol 2005;23:3227–3234.
Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 2005;128:270–279.
Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–4349.
Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42:1093–1103.
Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005;23:5357–5364.
Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005;2:e17.
Tamborini E, Bonadiman L, Greco A, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 2004;127:294–299.
Wakai T, Kanda T, Hirota S, et al. Late resistance to imatinib therapy in a metastatic gastrointestinal stromal tumour is associated with a second KIT mutation. Br J Cancer 2004;90:2059–2061.
Chen LL, Trent JC, Wu EF, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res 2004;64:5913–5919.
Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005;11:4182–4190.
Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res 2006;12:1743–1749.
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005;352:786–792.
Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2:e73.
Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA 2005;102:7665–7670.
Daub H, Specht K, Ullrich A . Strategies to overcome resistance to targeted protein kinase inhibitors. Nat Rev Drug Discov 2004;3:1001–1010.
McLean SR, Gana-Weisz M, Hartzoulakis B, et al. Imatinib binding and cKIT inhibition is abrogated by the cKIT kinase domain I missense mutation Val654Ala. Mol Cancer Ther 2005;4:2008–2015.
Valent P, Akin C, Sperr WR, et al. Mastocytosis: pathology, genetics, and current options for therapy. Leuk Lymphoma 2005;46:35–48.
Fletcher JA, Corless CL, Dimitrijevic S, et al. Mechanisms of resistance to imatinib mesylate (IM) in advanced gastrointestinal stromal tumor (GIST). Proc Am Soc Clin Oncol 2003;22:3275.
Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst 2005;97:643–655.
Elkind NB, Szentpetery Z, Apati A, et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib). Cancer Res 2005;65:1770–1777.
Sebolt-Leopold JS, English JM . Mechanisms of drug inhibition of signalling molecules. Nature 2006;441:457–462.
Schittenhelm MM, Shiraga S, Schroeder A, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res 2006;66:473–481.
Prenen H, Guetens G, de Boeck G, et al. Cellular uptake of the tyrosine kinase inhibitors imatinib and AMN107 in gastrointestinal stromal tumor cell lines. Pharmacology 2006;77:11–16.
Atkins M, Jones CA, Kirkpatrick P . Sunitinib maleate. Nat Rev Drug Discov 2006;5:279–280.
Carter TA, Wodicka LM, Shah NP, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA 2005;102:11011–11016.
Adrian FJ, Ding Q, Sim T, et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol 2006;2:95–102.
Whitesell L, Lindquist SL . HSP90 and the chaperoning of cancer. Nat Rev Cancer 2005;5:761–772.
Bauer S, Yu L, Read M, et al. IPI540, a novel HSP90 inhibitor, causes inhibition and degradation of KIT in imatinib-resistant GIST: rationale for therapeutic targeting in GIST. Clin Cancer Res 2005;11:91111S.
Shimamura T, Lowell AM, Engelman JA, et al. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res 2005;65:6401–6408.
Duensing A, Medeiros F, McConarty B, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 2004;23:3999–4006.
Van Oosterom A, Reichardt P, Blay JY, et al. A phase I/II trial of the oral mTOR-inhibitor everolimus (E) and imatinib mesylate (IM) in patients (pts) with gastrointestinal stromal tumor (GIST) refractory to IM: study update. J Clin Oncol 2005;23:824S.
Ihle NT, Paine-Murrieta G, Berggren MI, et al. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther 2005;4:1349–1357.
Janmaat ML, Rodriguez JA, Gallegos-Ruiz M, et al. Enhanced cytotoxicity induced by gefitinib and specific inhibitors of the Ras or phosphatidyl inositol-3 kinase pathways in non-small cell lung cancer cells. Int J Cancer 2006;118:209–214.
Donato NJ, Wu JY, Stapley J, et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood 2003;101:690–698.
Acknowledgements
We would like to thank Stefan Duensing for critically reading the manuscript. Work in AD's laboratory is supported by the GIST Cancer Research Fund (GCRF) and the numerous individuals who contributed through their donations.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Conflict of interest
The authors state no conflict of interest.
Rights and permissions
About this article
Cite this article
Rubin, B., Duensing, A. Mechanisms of resistance to small molecule kinase inhibition in the treatment of solid tumors. Lab Invest 86, 981–986 (2006). https://doi.org/10.1038/labinvest.3700466
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700466
Keywords
This article is cited by
-
Inhibition of USP28 overcomes Cisplatin-resistance of squamous tumors by suppression of the Fanconi anemia pathway
Cell Death & Differentiation (2022)
-
Oncogenic signaling by Kit tyrosine kinase occurs selectively on the Golgi apparatus in gastrointestinal stromal tumors
Oncogene (2017)
-
Dual inhibiting EGFR and VEGF pathways versus EGFR-TKIs alone in the treatment of advanced non-small-cell lung cancer: a meta-analysis of randomized controlled trials
Clinical and Translational Oncology (2016)
-
Clinical Implications of BMI-1 in Cancer Stem Cells of Laryngeal Carcinoma
Cell Biochemistry and Biophysics (2015)
-
Phase I study of weekly paclitaxel in combination with pazopanib and lapatinib in advanced solid malignancies
British Journal of Cancer (2014)
