
Abstract
Inflammatory bowel diseases (IBD) are chronic inflammatory disorders whose etiology remains unknown. Reports have shown that infiltration of leukocytes into intestinal tissue is a pathognomonic hallmark for this disease. Leukocyte β2 integrins are heterodimeric adhesion membrane proteins that are exclusively expressed on leukocytes and participate in immune cell adhesion and activation. In this study, we examined the pathophysiological role of the β2 integrins CD18, CD11a, and CD11b in the pathogenesis of dextran sodium sulfte (DSS)-induced experimental colitis. Disease activity was measured by daily assessment of clinical parameters including stool consistency, weight loss, occult blood, and gross rectal bleeding. Histopathological changes including severity of inflammation, surface epithelial/crypt damage, and depth of injury were also determined. The CD18 null and CD11a null mice had significantly lower disease activity and cumulative histopathological scores compared to wild-type mice. Interestingly, CD11b null mice did not show protection against DSS colitis and displayed increased disease activity compared to wild-type mice. Examination of specific leukocyte populations in the distal colon from various mice revealed significant attenuation of neutrophil and macrophage infiltrates in CD18, CD11a, and CD11b null mice. Surprisingly, the CD11b null mice showed a significant increase in plasma cell infiltration in response to DSS suggesting that this molecule may influence plasma cell function during colitis. This study demonstrates that genetic loss of CD18 or CD11a is protective during experimental colitis and that CD11b may serve a regulatory role during development of disease.
Similar content being viewed by others

Main
Inflammatory bowel diseases (IBD), including Crohn's disease and ulcerative colitis, are chronic inflammatory disorders of the gastrointestinal tract, which may also involve the small intestine that result in significant morbidity with limited therapeutic interventions. The exact causes of IBD are not known, and several factors have been reported to be involved in the development of disease, including immune dysfunction, genetic susceptibility, and bacterial flora within the intestinal environment.1, 2, 3 Although the specific causes of IBD are poorly understood, the pathological nature has been extensively defined and characterized by the presence of a granulomatous inflammatory process indicative of chronic inflammation.4, 5
The broad pathological process of IBD involves inflammation, ulceration, and subsequent regeneration of intestinal mucosa.6, 7, 8, 9 Thus, a central feature of IBD is a chronic immune dysregulation, which is the primary mediator of tissue damage associated with colitis.4, 5, 10 Several leukocyte types have been implicated in IBD pathogenesis, with T cells, neutrophils, and monocytes serving important roles in modulating chronic immune responses and subsequent tissue damage.11 However, the mechanisms regulating immune cell recruitment and function during IBD remain poorly understood.
Studies in several inflammatory models suggest that the leukocyte β2 integrins play important roles in the pathophysiology of several chronic and autoimmune disorders.12, 13, 14, 15, 16 The β2 integrins are heterodimeric proteins that consist of a common beta subunit (CD18) noncovalently attached to different alpha subunits (CD11a–d).17 The β2 integrin family, or CD18 integrins, consists of four different adhesion proteins LFA-1 (CD11a/CD18), Mac-1 (CD11b/CD18), p150/95 (CD11c/CD18), and CD11d/CD18. Importantly, expression of the β2 integrins is limited to leukocytes, which indicates their importance for immune responses. Members of the immunoglobulin superfamily are major ligands for the β2 integrins, including ICAM-1, 2, 3, and JAM-A and B.18, 19 Moreover, the β2 integrins are critical mediators of leukocyte recruitment, co-stimulation and activation, and leukocyte-mediated tissue remodeling. Data from our lab and others using mouse models of arthritis, SLE, psoriasis, and autoimmune diabetes show that the β2 integrins serve as the dominant adhesion molecules involved in chronic immune cell recruitment and activation, despite prominent expression of other known leukocyte adhesion proteins (eg VLA-4).12, 13, 15, 20 However, the pathological importance of β2 integrin regulation of leukocyte recruitment and function during experimental colitis is not known.
Several models of experimental colitis have been reported that display various pathophysiological aspects of human IBD.21, 22, 23 While no model serves as a complete surrogate for human disease, many salient clinical features can be examined depending on the method used to induce experimental colitis. The dextran sodium sulfate (DSS) model, originally reported by Okayasu et al,24 has been used extensively to investigate the role of various leukocytes during severe colitis. The clinical features of the DSS model includes weight loss, loose stool/diarrhea, and occult and gross rectal bleeding. Histopathological analysis typically reveals extensive crypt and epithelial cell damage, significant infiltration of granulocytes and mononuclear immune cells, tissue edema, and often frank ulceration.24, 25 Immunopathogenesis of the DSS model involves multiple immune cell types implicated in human IBD including neutrophils and monocytes, with lymphocytes serving a regulatory role.22, 25, 26, 27, 28 Therefore, we used the DSS colitis model to determine the pathological role of leukocyte β2 integrins during experimental colitis. Here, we report that gene targeted null deficiency of CD18 β2 integrin and CD11a (αL integrin) significantly attenuates the development of DSS-induced colitis, and that loss of CD11b (αM chain) actually enhanced DSS-induced colitis. These data suggest that targeted therapeutic intervention aimed at CD11a/CD18 may be useful in the treatment of IBD.
Materials and methods
Animals
Mice used for this study were bred and housed at the Louisiana State University Health Sciences Center-Shreveport animal resource facility and maintained according to the National Research Council guide for the care and use of laboratory animals. In total, 20 12-week-old male CD18 null−/−, CD11a null−/−, CD11b null−/−, or wild-type C57BL/6J mice were used for this study. These mice have been previously reported to be completely devoid of CD18, CD11a, or CD11b expression and were kindly provided by Dr Daniel Bullard from the University of Alabama at Birmingham.29, 30
3% Dextran Sodium Sulfate Induced Model of Colitis
DSS colitis was induced as previously reported.24, 25 Briefly, 3% DSS (MW=44 000) was added to the drinking water and available to the mice ad libitum. Water consumption was determined using graduated drinking bottles in order to determine the DSS load consumed to ensure that the minimal amount of DSS necessary to elicit disease was consumed.31 Over the 6-day study protocol, animals were monitored daily for weight loss, stool consistency, and occult and gross bleeding to obtain a clinical index of disease activity. On the morning of the 7th day, mice were anesthetized with 100 mg/kg ketamine and 8 mg/kg xylazene, and euthanized by cervical dislocation. The colon tissue was then removed and fixed in 10% buffered formalin. Tissue was then processed for routine H&E histopathology.
Assessment of Clinical Disease Activity
Clinical assessment of disease activity was performed using a validated scoring system previously reported.25, 32 Each animal was weighed daily to determine the percent change of weight during the DSS protocol. Fecal pellets were given a stool consistency score of 0 corresponding to a normal fecal pellet, 2 corresponding to a loose fecal pellet, or 4 corresponding to frank diarrhea. Occult blood was also scored using the Coloscreen occult blood card test and scored as 0 for negative and 2 for positive. Gross bleeding was determined as seen by wet blood on or around the anus (not dry blood in fecal material) and scored as 0 for negative and 4 for positive. A daily cumulative disease activity index (DAI) for each mouse was calculated using all of the above parameters.
Blood and Mesenteric Lymph Node Collection and Bacteriology
The thorax and abdomen of each mouse was shaved and scrubbed first with chlorhexidine and then with 70% ethanol. A 23 gauge, 1-inch needle was inserted caudal and right of the xiphoid process and directed towards the left shoulder until the heart was punctured. Blood was withdrawn and 0.5 ml was immediately placed into a BD (Sparks, MD, USA) Bactec Peds Plus/F blood culture vial. The abdominal wall was then incised along the linea alba to expose the viscera. The cecum was exteriorized onto a sterile drape to expose the mesenteric lymph nodes. The nodes and associated adipose tissue were aseptically removed and placed on PBS-moistened gauze in a sterile Petri dish. Tissue samples were homogenized in 0.5 ml of sterile saline using a small tissue grinder. A portion of the homogenized tissue sample was diluted 1:10 using sterile saline. A volume of 0.1 ml of the tissue and 0.1 ml of the 1:10 diluted tissue samples were plated to a chocolate agar plate (Remel Inc., Lenexa, KS, USA) and incubated at 35°C for 5 days. Blood cultures were processed using the automated 9240 BACTEC blood culture system (BD Diagnostics, Sparks, MD, USA). All blood and mesenteric lymph node cultures were performed and processed in the LSUHSC clinical microbiology laboratory.
Tissue Histopathology
Pathological scoring was performed on distal segments of the colon in a blinded manner using a validated scoring system.25, 32 Hematoxylin and eosin histopathology was scored based on three parameters: severity of inflammation (0–3; none, slight, moderate, severe), depth of injury (0–3; none, mucosal, mucosal and submucosal, and transmural), and crypt damage (0–4; none, basal ⅓ damaged, basal ⅔ damaged, only surface epithelium intact, loss of entire crypt and epithelium). Each parameter score was multiplied by a factor reflecting percent involvement of the intestinal wall ( × 1: 0–25%, × 2: 26–50%, × 3: 51–75%, × 4: 76–100%) and summed to obtain a histopathology score (maximum of 40 possible). The number of neutrophils, lymphocytes, and plasma cells were also counted per 40 × field. Six sections were scored per animal and averaged. The resulting data were statistically compared using a one-way ANOVA with Bonferroni's post-testing. A value of P<0.05 was required to achieve statistical significance. All data are reported as the mean±s.e.
Macrophage Staining
Tissue macrophage staining was performed as previously reported.12 Rat anti-mouse F4/80 primary antibody was used to stain tissue macrophages using the automated DakoCytomation stainer. Briefly, 10 μm formalin fixed sections were blocked, incubated with the primary antibody (1:60 dilution) followed by secondary antibody incubation, and developed using DAB reagent. Sections were counterstained with hematoxylin and mounted. The number of positive F4/80 staining leukocytes was counted per 400 × field. Six sections were scored per animal and averaged. The resulting data were compared using one-way ANOVA with Bonferroni's post-testing. A value of P<0.05 was required for statistical significance. All data are reported as the mean±s.e.
Results
Clinical Features of DSS Induced Colitis
Clinical features of DSS colitis were determined by daily monitoring of weight change, stool consistency, occult, and gross bleeding. Figure 1 illustrates the various clinical parameters of the different genetic mutant mice during DSS treatment. All vehicle control (ie regular water) mice of each genotype were negative for any clinical features of colitis (ie score of 0) and therefore were not graphed in the following panels. Panel a illustrates the percent weight change among the different genotypes. Wild type and CD11b null mice showed a substantial weight loss whereas CD18 null and CD11a null mice displayed a significantly smaller amount of weight loss. Panel b shows that all of the genotype groups displayed progressively higher stool scores, with the CD11a null mice showing significantly lower stool scores than wild-type mice. Panel c reports the presence of occult blood as determined by the Coloscreen occult blood test. Both the CD18 and CD11a null mice show a significant temporal delay in occult bleeding compared to wild-type mice. No significant difference was seen between wild type and CD11b even though CD11b null mice had a trend to have more blood in their stools. Mice were also observed for gross bleeding as seen by wet blood on or around the anus (not dry blood in fecal material). Panel d shows that wild-type mice typically displayed gross bleeding between days 5 and 6. However, CD18 null and CD11a null mice showed a significant time delay in gross bleeding onset. Only one CD18 null and three CD11a null mice showing gross bleeding compared to seven of the wild-type mice. Interestingly, gross bleeding occurred earlier and was seen in all CD11b null mice compared to wild-type mice.

Clinical symptoms of DSS colitis—clinical symptoms of DSS experimental colitis were measured in wild type (▪), CD18 null (▴), CD11a null (▾), and CD11b null (•) mice. (a–d) Percent weight change, stool score, occult blood score, and gross bleeding score, respectively. *P<0.05 vs wild-type mice.
A DAI was calculated for each mouse using measurements from the four clinical parameters presented in Figure 1 with weight loss, gross bleeding, stool consistency, and occult blood contributing to the cumulative score. Figure 2 shows that the cumulative DAI of wild-type mice begins to increase between days 3 and 4. Interestingly, the CD11b DAI is significantly greater than that of the wild-type mice at day 3 and remains greater throughout the study period. CD18 null DAI was significantly blunted overall and was temporally delayed compared to that of the wild-type mice. Lastly, CD11a null DAI was significantly attenuated for the first 4 days, but became equivalent to wild type levels between days 5 and 6, primarily due to the later gross bleeding. Together, these data demonstrate that genetic loss of all β2 integrins confers protection against DSS colitis, whereas loss of CD11a attenuates and loss of CD11b enhances clinical symptoms of experimental colitis.

DSS-induced cumulative disease activity index—a cumulative disease activity index was calculated using clinical symptom data for each time point for wild type (▪), CD18 null (▴), CD11a null (▾), and CD11b null (•) mice. *P<0.05 increased compared to wild-type mice. #P<0.05 decreased compared to wild-type mice.
Histopathology of DSS Induced Colitis
The microscopic sections of the distal colon from the different mouse genotypes showed a wide range of histopathologic features, which included edema, epithelial cell and crypt damage, and inflammatory infiltrates (Figure 3). The degree of severity of the edema was graded according to the following three criteria: (1) severe—when the space between the muscularis mucosa and the muscularis propria as a result of edema fluid collection was equal to or greater than the thickness of the mucosa; (2) moderate—when the space was between 30 and 100% of the mucosal thickness; and (3) mild—when the space was less than 30% of the mucosal thickness. The predominant histopathologic changes found in distal colon segments of wild-type mice (ie control genotype) after DSS consumption included (1) diffuse mucosal and sub mucosal edema in all levels of the histological sections; (2) surface epithelial damage and crypt drop out; and (3) abundant inflammatory cell infiltrates. The most striking histopathologic changes in the distal colon from wild-type mice were surface epithelial damage and mucosal crypt dropout. This pathologic change ranged from moderate to severe and was identified in the distal colons from all 10 of the wild-type mice. Full thickness loss of the mucosal surface including crypt damage was noted in six of the 10 specimens. Interestingly, in addition to the acute inflammatory cell infiltrates noted in the distal colon, a mixture of chronic inflammatory cells including plasma cell and lymphocytes were recognized as well. In contrast, the distal colons from all of the CD18 null mice after DSS consumption had completely intact surface epithelium including mucosal crypts. The only histopathological feature noted was mild submucosal edema, which was free of inflammatory infiltrates. These results demonstrate that loss of all four β2 integrins significantly protects the distal colon from DSS mediated colitis.

DSS histopathology of the distal colon. (a–d) H&E histopathology for DSS treated wild type, CD18 null, CD11a null, CD11b null mice. Conversely, (e) illustrates normal histomorphology of untreated wild-type mice. Photomicrographs were taken at × 200.
We then examined the role of individual β2 integrins using CD11a null (ie LFA-1 null) mice and CD11b null (ie Mac-1 null) mice in the DSS model. Microscopic sections of distal colon segments from the CD11a null mice after DSS consumption had histopathological changes consistent with an inflammatory process including (1) edema, (2) epithelial damage, and (3) acute inflammatory infiltrates. However, all three histopathological features were less severe in DSS treated CD11a null colons than in wild-type colons, and a large portion of the distal colons were entirely normal with no evidence of inflammatory changes. Submucosal edema was noted in a few areas and was largely very mild in contrast to that seen in the wild-type mice. The pattern of surface epithelial damage found in the distal colons from the CD11a null mice was unlike that found in the distal colons from the wild-type mice in that a smaller percentage of the mucosal surface area was affected. Six of the CD11a null mice had 15% of the surface area affected, three had 20% of the surface area affected, and one had 35% of the surface area affected compared to ≥40% of the luminal surface area affected in all wild-type mice. Moreover, when epithelial cell damage was observed in the CD11a null mice it was confined to the basal two/thirds of the mucosal surface, rather than the full thickness involvement seen in the wild-type mice.
In contrast to the CD18 and CD11a null mice, the histopathologic changes in distal colons of the CD11b null mice were more similar to the changes observed in the colons of the wild-type mice and ranged from moderate to severe. The most striking histopathologic feature was surface epithelial damage and mucosal crypt dropout. Full thickness loss of the mucosal surface including crypt damage was noted in the colons from 80% of the CD11b null mice. In total, 80% of the CD11b null specimens also had 100% surface area damage with the remaining 20% showing 40% surface epithelial damage. However, unlike the wild-type mice, the pattern of involvement in the distal colons of CD11b null mice was predominantly patchy. We found that full thickness surface epithelial damage would involve 5–15% of the surface area in some of the microscopic sections with 100% surface area in other sections of the same distal colonic segment.
Cumulative DSS histopathological scores calculated by evaluating several tissue pathology parameters comparing the different genotypes is shown in Figure 4. As suggested from the DAI and histopathology, CD18 null mice were completely protected from DSS mediated colitis. The histopathology score of CD11a null mice was also significantly decreased compared to wild-type mice, but was still greater than that of the CD18 null mice. Surprisingly, the histopathology score of CD11b null mice was slightly but not significantly greater than DSS treated wild-type mice. Together, these data suggest that CD18 and CD11a play important roles in mediating tissue damage in response to DSS treatment.

DSS-mediated cumulative histopathology score—DSS mediated tissue damage was determined using the histopathology scoring system as described in the Materials and methods. CD18 null and CD11a null mice show significant attenuation of DSS-mediated histopathology. *P<0.05 compared to wild type.
Loss of β2 Integrins does not Alter Bacterial Translocation from the Gut
Leukocyte β2 integrins, namely CD11b, have been reported to be important for defense against infective agents and facilitate bacterial clearance.17, 30 Therefore, we next examined whether genetic deficiency of β2 integrins might affect bacterial translocation from the gut, thereby exacerbating tissue pathology as seen in CD11b null mice. Table 1 reports bacteriology results from blood and mesenteric lymph node cultures from all of the various mice used in this study. Blood cultures were negative among all of the genetic variations, while one out of four mice showed minimal growth of routine organisms from mesenteric lymph nodes. Together, these data demonstrate that differential susceptibility to DSS pathology among the various mutants does not involve an increase in bacterial burden.
Loss of β2 Integrins Differentially Affects DSS Mediated Leukocyte Recruitment
β2 Integrins have been shown to be important in regulating immune cell adhesion and recruitment in several inflammation and chronic inflammatory disease models.12, 14, 15, 16, 17 Therefore, we determined the numbers of neutrophils, lymphocytes, and plasma cells per × 400 field in the colons of each of the genotypes. Figure 5a reports the numbers of infiltrating neutrophils. The colons of the CD18 null mice showed the fewest numbers of neutrophils followed by the colons of CD11a null mice. Interestingly, the colons of CD11b null mice did show a small but statistically significant decrease in neutrophil infiltrates. Figure 5b demonstrates that lymphocyte infiltration was not altered in the colons of CD18 null, CD11a null, and CD11b null mice compared to those of the wild-type mice. Lastly, c reports the number of plasma cells in the colons comparing the various genotypes. Interestingly, the colons of CD11b null mice had a significant two-fold increase in plasma cell numbers in response to DSS treatment. Together, these data demonstrate that CD18 and CD11a are primarily important for neutrophil infiltration during DSS colitis, and that CD11b may be important in regulating the numbers of distal colon plasma cells during experimental colitis.

DSS stimulated leukocyte infiltrates—leukocyte infiltration in response to DSS treatment was determined for all genotypes. (a–c) Number of neutrophils, lymphocytes, or plasma cells per × 400 field, respectively. *P<0.05 genotype DSS vs genotype control, #P<0.01 wild-type DSS vs genetic mutant DSS, ⋀P<0.05 wild-type DSS vs CD11b null DSS.
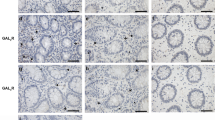
A previous report by Kringlestein et al25 has shown that macrophages play a key pathophysiological role during DSS induced colitis. Therefore, we examined whether genetic deficiency of β2 integrins alters macrophage infiltration in response to DSS. Figure 6 shows macrophage F4/80 staining in wild type, CD18 null, CD11a null, and CD11b null DSS treated colons, respectively (Figure 6a–d). Further quantification of macrophage numbers per × 400 field shows that loss of β2 integrin expression significantly attenuates macrophage infiltration in response to DSS (Figure 6e). These data are consistent with the previous finding that macrophage infiltration contributes to DSS histopathology and clearly reveals that β2 integrins play a dominant role in regulating tissue macrophage accumulation.

DSS-mediated macrophage infiltration—tissue macrophages were identified based on F4/80 antigen staining. (a–d) Macrophage infiltration in wild type, CD18 null, CD11a null, or CD11b null DSS treated colons, respectively. Arrows indicate positive staining cells. Panel e reports the number of macrophages per × 400 field revealing a significant attenuation of macrophage staining in all of the β2 integrin deficient mice. *P<0.01 wild-type control vs wild-type DSS, #P<0.01 wild-type DSS vs genetic mutant DSS.
Discussion
Immune system dysregulation and increased leukocyte recruitment are key pathological features of both human IBD and experimental colitis models.33, 34 Much effort has been focused on determining specific molecular mechanisms responsible for immune cell mediated tissue damage during colitis. Several reports have previously identified leukocyte and endothelial cell adhesion molecules as potentially important regulators of leukocyte recruitment in human IBD and experimental colitis.35, 36 Studies have implicated endothelial cell ICAM-1 and MadCAM-1 along with leukocyte α4β1 (VLA-4) and α4β7 expression as primary mediators of leukocyte recruitment in colitis.37, 38, 39, 40 However, other reports have clearly documented increased expression of β2 integrins, including LFA-1 (CD11a/CD18) and Mac-1 (CD11b/CD18), in Crohn's disease and ulcerative colitis suggesting that these molecules may also play an important role in governing chronic inflammation of the bowel.41, 42, 43, 44 Few studies have directly examined the role of these molecules during experimental colitis with these reports documenting modification of trinitrobenzene sulfonic acid (TNBS) colitis with anti-CD18 or CD11b/CD18 antibodies.45, 46 We report here that the β2 integrin LFA-1 (CD11a/CD18) plays an important pathological role, whereas Mac-1 (CD11b/CD18) provides a critical regulatory role in DSS-induced experimental colitis. Several possible explanations could account for the difference in outcome regarding CD11b. First, the model used in the antibody blockade study was the TNBS colitis model, which may have a differential requirement for CD11b expression for leukocyte-mediated pathology. Second, due to a global gene targeted mutation, the genetic loss of CD11b may have more far reaching consequences on other leukocyte populations and functions, for example intracellular signaling, compared to monoclonal antibody treatment. Lastly, previous reports using CD11b blocking monoclonal antibodies have shown that these antibodies can bind to and activate Fc receptors which may act to alter immune responses.47 Nonetheless, our data reveal a dichotomous role for different members of the β2 integrin family in regulating DSS experimental colitis.
Previous studies have shown that DSS mediated colitis is largely due to increased neutrophil and monocyte recruitment, such that anti-neutrophil serum or blockade of monocyte infiltration significantly attenuates disease.25, 27 Our data also demonstrate that neutrophil recruitment is important for DSS colitis and that CD18 and CD11a play key roles in facilitating this process. This observation is surprising since many studies have reported that CD11b/CD18 (Mac-1) serves as the primary mediator of neutrophil–endothelial cell interactions and subsequent recruitment.48, 49 While we did see a small attenuation of neutrophil infiltration in the CD11b null mouse, the amount of inhibition was not remotely similar to the degree of protection conferred in CD18 or CD11a null mice. Aside from the fact that loss of CD11a attenuates neutrophil firm adhesion, another possible explanation for our observations may lie in the recent understanding that CD11a/CD18 (LFA-1) serves to stabilize leukocyte slow rolling, a critical event in facilitating leukocyte activation and subsequent firm adhesion.50 Moreover, it appears that CD11b/CD18 primarily uses endothelial cell ICAM-1 in facilitating leukocyte adhesion; whereas, CD11a/CD18 binds to other ligands besides ICAM-1, including ICAM-2 and JAM-A.17, 18, 51 Therefore, it is likely that loss of CD11a/CD18 eliminates several adhesion pathways that may be necessary for neutrophil recruitment during colitis, which remain intact and facilitate neutrophil recruitment in CD11b null mice, thereby explaining minimal to small defects in neutrophil recruitment in CD11b null distal colons. Future studies are necessary to better understand differential effects of CD11a and CD11b expression in regulating neutrophil recruitment during DSS colitis.
CD11b/CD18 has been reported to play an important role in regulating neutrophil adhesion, transmigration, and subsequent tissue damage through cellular activation, release of intracellular enzymes, and generation of reactive oxygen species.49, 52 Therefore, it is surprising that CD11b null mice display enhanced DSS colitis. A previous report by Coxon et al53 has shown that CD11b plays a role in regulating neutrophil apoptosis such that genetic loss of CD11b delays apoptosis thereby possibly increasing the capacity for tissue damage. However, the contribution of such a response is likely minimal as we did not observe an increase in neutrophil infiltrates as might be expected with a defect in apoptosis. CD11b is also important for innate immune/host defense responses in that loss of CD11b significantly impairs resistance against bacterial challenge.54, 55 Moreover, it has been reported that the severity of DSS colitis is influenced by enteric bacterial; therefore, it could be that genetic loss of CD11b is permissive for bacterial translocation from the colon thereby enhancing disease.56, 57 Blood and mesenteric lymph node cultures from all of the genetic variants in this study were not different from one another, suggesting that bacterial burden is not a mitigating factor for enhanced disease activity in CD11b null mice.
It is possible that loss of CD11b may affect other leukocyte populations that influence the progression of colitis. Indeed, our data demonstrate a significant increase in the numbers of plasma cells in colitic tissue of CD11b null mice. Previous reports have implicated a role for B cells during DSS colitis, yet the true pathological importance of this cell population during experimental colitis remains poorly understood.58, 59 CD11b may be important for B-cell function and/or recruitment. We have recently reported that the loss of CD11b in the MRL/MpJ-Faslpr mouse lupus model results in enhanced lymphadenopathy with focal accumulation of plasma and Mott cells.15 A recent study has also shown that CD11b may also govern T-cell development and function, in that the loss of CD11b decreased CD3 and CD28 expression and CD4/CD8 ratios.60 Early reports dismissed a role for T cells in DSS colitis; however, recent reports suggest that this cell population may participate in a regulatory manner through influencing recruitment of specific leukocyte populations (ie monocytes vs granulocytes) or by providing growth factors necessary for epithelial cell survival and regeneration during DSS colitis.26, 28 Therefore, it is possible that CD11b deficiency may alter T-cell regulatory functions during DSS colitis. While both of these possibilities may be involved, future studies are needed to better understand how CD11b modulates DSS induced colitis.
Much attention has been focused on the role of α4 integrin in IBD and experimental colitis, with the blocking agent Nataluzimab currently under evaluation for IBD treatment.61, 62 It has also recently been reported that Natalizumab therapy may result in undesirable complications such as multifocal leukoencephalopathy.63 In this study, we have shown that CD11a/CD18 plays an important pathological role governing DSS experimental colitis through regulating leukocyte infiltration and subsequent tissue damage. Importantly, the anti-CD11a blocking antibody Efalizumab is currently approved for the treatment of psoriasis with several other novel small molecule CD11a antagonists currently under development.64, 65, 66, 67, 68 Our findings together with previous reports of CD11a and CD18 expression in human IBD specimens suggest that targeting of these molecules may provide another therapeutic treatment for IBD.
References
Mahida YR, Rolfe VE . Host–bacterial interactions in inflammatory bowel disease. Clin Sci (Lond) 2004;107:331–341.
Rioux JD . Progress towards identifying inflammatory bowel disease susceptibility genes. Novartis Found Symp 2004;263:3–11; discussion 11–16, 211–218.
Powrie F . Immune regulation in the intestine: a balancing act between effector and regulatory T cell responses. Ann NY Acad Sci 2004;1029:132–141.
Kusugami K, Ina K, Ando T, et al. Immunomodulatory therapy for inflammatory bowel disease. J Gastroenterol 2004;39:1129–1137.
Wen Z, Fiocchi C . Inflammatory bowel disease: autoimmune or immune-mediated pathogenesis? Clin Dev Immunol 2004;11:195–204.
Farthing MJ . Severe inflammatory bowel disease: medical management. Dig Dis 2003;21:46–53.
Ardizzone S, Porro GB . Inflammatory bowel disease: new insights into pathogenesis and treatment. J Intern Med 2002;252:475–496.
Abreu MT . The pathogenesis of inflammatory bowel disease: translational implications for clinicians. Curr Gastroenterol Rep 2002;4:481–489.
Hommes DW, van Deventer SJ . Endoscopy in inflammatory bowel diseases. Gastroenterology 2004;126:1561–1573.
Panes J . Inflammatory bowel disease: pathogenesis and targets for therapeutic interventions. Acta Physiol Scand 2001;173:159–165.
Sartor RB . Pathogenesis and immune mechanisms of chronic inflammatory bowel diseases. Am J Gastroenterol 1997;92:5S–11S.
Barlow SC, Langston W, Matthews KM, et al. CD18 deficiency protects against multiple low-dose streptozotocin-induced diabetes. Am J Pathol 2004;165:1849–1852.
Barlow SC, Collins RG, Ball NJ, et al. Psoriasiform dermatitis susceptibility in Itgb2(tm1Bay) PL/J mice requires low-level CD18 expression and at least two additional loci for progression to severe disease. Am J Pathol 2003;163:197–202.
Kevil CG, Bullard DC . Roles of leukocyte/endothelial cell adhesion molecules in the pathogenesis of vasculitis. Am J Med 1999;106:677–687.
Kevil CG, Hicks MJ, He X, et al. Loss of LFA-1, but not Mac-1, protects MRL/MpJ-Fas(lpr) mice from autoimmune disease. Am J Pathol 2004;165:609–616.
Forlow SB, White EJ, Barlow SC, et al. Severe inflammatory defect and reduced viability in CD18 and E-selectin double-mutant mice. J Clin Invest 2000;106:1457–1466.
Harris ES, McIntyre TM, Prescott SM, et al. The leukocyte integrins. J Biol Chem 2000;275:23409–23412.
Kevil CG . Endothelial cell activation in inflammation: lessons from mutant mouse models. Pathophysiology 2003;9:63–74.
Chavakis T, Preissner KT, Santoso S . Leukocyte trans-endothelial migration: JAMs add new pieces to the puzzle. Thromb Haemost 2003;89:13–17.
Watts GM, Beurskens FJ, Martin-Padura I, et al. Manifestations of inflammatory arthritis are critically dependent on LFA-1. J Immunol 2005;174:3668–3675.
Elson CO, Sartor RB, Tennyson GS, et al. Experimental models of inflammatory bowel disease. Gastroenterology 1995;109:1344–1367.
Elson CO, Cong Y, Brandwein S, et al. Experimental models to study molecular mechanisms underlying intestinal inflammation. Ann NY Acad Sci 1998;859:85–95.
Pizarro TT, Arseneau KO, Bamias G, et al. Mouse models for the study of Crohn's disease. Trends Mol Med 2003;9:218–222.
Okayasu I, Hatakeyama S, Yamada M, et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990;98:694–702.
Krieglstein CF, Cerwinka WH, Sprague AG, et al. Collagen-binding integrin alpha1beta1 regulates intestinal inflammation in experimental colitis. J Clin Invest 2002;110:1773–1782.
Chen Y, Chou K, Fuchs E, et al. Protection of the intestinal mucosa by intraepithelial gamma delta T cells. Proc Natl Acad Sci USA 2002;99:14338–14343.
Domek MJ, Iwata F, Blackman EL, et al. Anti-neutrophil serum attenuates dextran sulfate sodium-induced colonic damage in the rat. Scand J Gastroenterol 1995;30:1089–1094.
Tsuchiya T, Fukuda S, Hamada H, et al. Role of gamma delta T cells in the inflammatory response of experimental colitis mice. J Immunol 2003;171:5507–5513.
Ding ZM, Babensee JE, Simon SI, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol 1999;163:5029–5038.
Scharffetter-Kochanek K, Lu H, Norman K, et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med 1998;188:119–131.
Vowinkel T, Kalogeris TJ, Mori M, et al. Impact of dextran sulfate sodium load on the severity of inflammation in experimental colitis. Dig Dis Sci 2004;49:556–564.
Dieleman LA, Ridwan BU, Tennyson GS, et al. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 1994;107:1643–1652.
Eksteen B, Miles AE, Grant AJ, et al. Lymphocyte homing in the pathogenesis of extra-intestinal manifestations of inflammatory bowel disease. Clin Med 2004;4:173–180.
Laroux FS, Grisham MB . Immunological basis of inflammatory bowel disease: role of the microcirculation. Microcirculation 2001;8:283–301.
Vainer B . Role of cell adhesion molecules in inflammatory bowel diseases. Scand J Gastroenterol 1997;32:401–410.
Vainer B, Nielsen OH, Horn T . Comparative studies of the colonic in situ expression of intercellular adhesion molecules (ICAM-1, -2, and -3), beta2 integrins (LFA-1, Mac-1, and p150,95), and PECAM-1 in ulcerative colitis and Crohn's disease. Am J Surg Pathol 2000;24:1115–1124.
Connor EM, Eppihimer MJ, Morise Z, et al. Expression of mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in acute and chronic inflammation. J Leukoc Biol 1999;65:349–355.
Papa A, Danese S, Urgesi R, et al. Intercellular adhesion molecule 1 gene polymorphisms in inflammatory bowel disease. Eur Rev Med Pharmacol Sci 2004;8:187–191.
Meenan J, Spaans J, Grool TA, et al. Altered expression of alpha 4 beta 7, a gut homing integrin, by circulating and mucosal T cells in colonic mucosal inflammation. Gut 1997;40:241–246.
Podolsky DK, Lobb R, King N, et al. Attenuation of colitis in the cotton-top tamarin by anti-alpha 4 integrin monoclonal antibody. J Clin Invest 1993;92:372–380.
Bernstein CN, Sargent M, Rector E . Alteration in expression of beta 2 integrins on lamina propria lymphocytes in ulcerative colitis and Crohn's disease. Clin Immunol 2002;104:67–72.
Bernstein CN, Sargent M, Gallatin WM . Beta2 integrin/ICAM expression in Crohn's disease. Clin Immunol Immunopathol 1998;86:147–160.
Bernstein CN, Sargent M, Rawsthorne P, et al. Peripheral blood lymphocyte beta 2 integrin and ICAM expression in inflammatory bowel disease. Dig Dis Sci 1997;42:2338–2349.
Kirman I, Nielsen OH . LFA-1 subunit expression in ulcerative colitis patients. Dig Dis Sci 1996;41:670–676.
Palmen MJ, Dijkstra CD, van der Ende MB, et al. Anti-CD11b/CD18 antibodies reduce inflammation in acute colitis in rats. Clin Exp Immunol 1995;101:351–356.
Wallace JL, Higa A, McKnight GW, et al. Prevention and reversal of experimental colitis by a monoclonal antibody which inhibits leukocyte adherence. Inflammation 1992;16:343–354.
Gadd SJ, Eher R, Majdic O, et al. Signal transduction via Fc gamma R and Mac-1 alpha-chain in monocytes and polymorphonuclear leucocytes. Immunology 1994; 81:611–617.
Li Z . The alphaMbeta2 integrin and its role in neutrophil function. Cell Res 1999;9:171–178.
Ross GD . Regulation of the adhesion vs cytotoxic functions of the Mac-1/CR3/alphaMbeta2-integrin glycoprotein. Crit Rev Immunol 2000;20:197–222.
Dunne JL, Ballantyne CM, Beaudet AL, et al. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood 2002;99:336–341.
Dunne JL, Collins RG, Beaudet AL, et al. Mac-1, but not LFA-1, uses intercellular adhesion molecule-1 to mediate slow leukocyte rolling in TNF-alpha-induced inflammation. J Immunol 2003;171:6105–6111.
van Spriel AB, Leusen JH, van Egmond M, et al. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood 2001;97:2478–2486.
Coxon A, Rieu P, Barkalow FJ, et al. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 1996;5:653–666.
Rosenkranz AR, Coxon A, Maurer M, et al. Impaired mast cell development and innate immunity in Mac-1 (CD11b/CD18, CR3)-deficient mice. J Immunol 1998;161:6463–6467.
Prince JE, Brayton CF, Fossett MC, et al. The differential roles of LFA-1 and Mac-1 in host defense against systemic infection with Streptococcus pneumoniae. J Immunol 2001;166:7362–7369.
Rath HC, Schultz M, Freitag R, et al. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect Immun 2001;69:2277–2285.
Hernandez GA, Appleyard CB . Bacterial load in animal models of acute and chronic ‘reactivated’ colitis. Digestion 2003;67:161–169.
Stevceva L, Pavli P, Husband AJ, et al. The inflammatory infiltrate in the acute stage of the dextran sulphate sodium induced colitis: B cell response differs depending on the percentage of DSS used to induce it. BMC Clin Pathol 2001;1:3.
Teramoto K, Miura S, Tsuzuki Y, et al. Increased lymphocyte trafficking to colonic microvessels is dependent on MAdCAM-1 and C-C chemokine mLARC/CCL20 in DSS-induced mice colitis. Clin Exp Immunol 2005;139:421–428.
Wu H, Rodgers JR, Perrard XY, et al. Deficiency of CD11b or CD11d results in reduced staphylococcal enterotoxin-induced T cell response and T cell phenotypic changes. J Immunol 2004;173:297–306.
Huryn DM, Ashwell S, Baudy R, et al. Synthesis, characterization and evaluation of pro-drugs of VLA-4 antagonists. Bioorg Med Chem Lett 2004;14:1651–1654.
Michetti P, Mottet C, Juillerat P, et al. Severe and steroid-resistant Crohn's disease. Digestion 2005;71:19–25.
Berger JR, Koralnik IJ . Progressive multifocal leukoencephalopathy and natalizumab—unforeseen consequences. N Engl J Med 2005;353:414–416.
Cather JC, Menter A . Efalizumab: continuous therapy for chronic psoriasis. Expert Opin Biol Ther 2005;5:393–403.
Huang M, Matthews K, Siahaan TJ, et al. Alpha L-integrin I domain cyclic peptide antagonist selectively inhibits T cell adhesion to pancreatic islet microvascular endothelium. Am J Physiol Gastrointest Liver Physiol 2005;288:G67–G73.
Wattanasin S, Kallen J, Myers S, et al. 1,4-Diazepane-2,5-diones as novel inhibitors of LFA-1. Bioorg Med Chem Lett 2005;15:1217–1220.
Weitz-Schmidt G, Welzenbach K, Dawson J, et al. Improved lymphocyte function-associated antigen-1 (LFA-1) inhibition by statin derivatives: molecular basis determined by X-ray analysis and monitoring of LFA-1 conformational changes in vitro and ex vivo. J Biol Chem 2004;279:46764–46771.
Yusuf-Makagiansar H, Anderson ME, Yakovleva TV, et al. Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as a therapeutic approach to inflammation and autoimmune diseases. Med Res Rev 2002;22:146–167.
Acknowledgements
We thank Mr Lamar Jones and Mrs Joyce Jackson for expert technical assistance and support from LSUHSC-S Center of Excellence for Arthritis and Rheumatology and NIH Grant DK 43785.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abdelbaqi, M., Chidlow, J., Matthews, K. et al. Regulation of dextran sodium sulfate induced colitis by leukocyte beta 2 integrins. Lab Invest 86, 380–390 (2006). https://doi.org/10.1038/labinvest.3700398
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700398
Keywords
This article is cited by
-
Mycobacterium bovis BCG increase the selected determinants of monocyte/macrophage activity, which were diminished in response to gastric pathogen Helicobacter pylori
Scientific Reports (2023)
-
Extracellular ATP mediates inflammatory responses in colitis via P2 × 7 receptor signaling
Scientific Reports (2016)
-
Immune/Inflammatory Response and Hypocontractility of Rabbit Colonic Smooth Muscle After TNBS-Induced Colitis
Digestive Diseases and Sciences (2016)
-
Loss of beta2-integrin-mediated cytoskeletal linkage reprogrammes dendritic cells to a mature migratory phenotype
Nature Communications (2014)
-
Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b
Nature Immunology (2010)
