
Abstract
Increased proliferation, defective apoptosis, and cytokine dysregulation of T lymphocytes are thought to be important for the pathogenesis of inflammatory bowel disease. Since these phenomena can be corrected by αCD2 mAb, we asked whether CD2 directed immunotherapy safely prevents and/or ameliorates adoptive transfer colitis. Colitis was induced by transfer of CD4+ T cell blasts to syngenic RAG1−/− mice or CD45RBhigh CD4+ T cells to SCID mice. The αCD2 mAb 12-15 or rat IgG was given, starting either initially or upon first signs of colitis. Disease activity was assessed by clinical monitoring, microscopic scoring, hemoccult, endoscopy, and blood count analysis. Cytokine production of stimulated LPL was measured by ELISA and cell proliferation by [3H]-thymidine incorporation. Parasite control was analyzed in a murine model of infection with Toxoplasma gondii. The αCD2 mAb significantly increased mean survival time when starting at transfer of blasts (survival >35 days: αCD2 69% vs 0% of controls, P<0.001). In the SCID colitis model hematochezia and macroscopic colitis were delayed. When used in established T-cell blast colitis, the benefit was less pronounced, even in combination with dexamethasone (mean survival±s.e.m.: αCD2+dexa: 13.5±2.9 vs dexa+IgG: 6.3±1.0, P<0.05). In the preventive experiment the αCD2 mAb markedly reduced IL-2 secretion and T-cell proliferation. The immune response towards Toxoplasma gondii was not impaired. These studies show for the first time that CD2 directed immunotherapy can attenuate or delay adoptive transfer colitis and ameliorate established colitis. Most likely inhibition of IL-2 secretion and T-cell proliferation are responsible for these effects. Still, immune defence towards T. gondii is maintained.
Similar content being viewed by others


Main
Inflammatory bowel disease (IBD) is characterized by mononuclear cell infiltration of affected bowel segments, consisting primarily of macrophages and T helper (Th) cells. The number of T helper cells can increase within the intestine by enhanced T-cell proliferation, reduced T-cell apoptosis, and increased adhesion.1, 2, 3, 4, 5 Proinflammatory cytokines are strongly involved in these processes showing a Th1 dominance in most animal models.6, 7, 8
CD2, a widely distributed glycoprotein, plays a pivotal role in the immunological synapse adapting T cells with antigen presenting cells.9, 10 Signals via CD2 are important for both antigen-dependent as well as antigen-independent T-cell activation.11, 12 For mucosal immunology, CD2 plays an even more important role, since lamina propria T cells (LPL-T) proliferate via CD2 but much less via the CD3/T-cell receptor complex, both in vitro and in vivo.13, 14 In addition, cytokine secretion by LPL-T as well as LPL-T-cell apoptosis is mediated via CD2.15, 16, 17 Also, CD2 seems to be involved in the induction of regulatory T cells.18, 19 MAb directed at CD2 inhibit T-cell proliferation,20, 21 induce LPL-T apoptosis,16 inhibit Th1 cytokine production, and induce Th2 and Th3 cytokines.22, 23, 24 Accordingly, αCD2 mAb were used successfully in both transplantation models,25, 26, 27, 28 arthritis and multiple sclerosis animal models.29, 30, 31 Clinical studies suggest that CD2 is a promising target in psoriasis.32 Therefore, we asked whether αCD2 mAb are effective in a colitis animal model, that is, adoptive transfer colitis. Specific immunotherapies (eg αTNF-α mAb) have been associated with impaired host defence.33 To investigate whether αCD2 mAb treatment impairs host defences against intracellular microorganisms, we analyzed numbers of parasites following oral infection of mice with Toxoplasma gondii. Following peroral infection of C57BL/6 mice with T. gondii, parasites replicate in the small intestines and induce a Th1-mediated small-intestinal immunopathology.34
Here, we demonstrate that αCD2 mAb treatment partially prevents transfer colitis and slows down the inflammatory course in established disease. At the same time, treatment appeared to be safe, since the host defence against T. gondii was not impaired.
Materials and methods
Animals
Donor mice (C57BL/6) and RAG1−/− mice were obtained from I. Förster, Munich, Germany, and were bred under specific pathogen-free (SPF) conditions at the Research Institute for Experimental Medicine (FEM), Berlin, Germany, or at the animal care facility of the University Hospital of Saarland at Homburg, Germany. BALB/c donor mice and C.B-17 SCID mice were obtained from Harlan, USA. Mice were kept in polycarbonate cages and had free access to sterile standard chow and water. All animal experiments were approved by the local animal protection committee.
Induction of Transfer Colitis
Two different models of transfer colitis were used: (1) Induction of colitis by CD4+ T cell blasts (‘T cell blast transfer colitis’) and (2) Induction of colitis by transfer of CD45RBhigh T cells (‘classical CD45RBhigh SCID transfer colitis’). For transfer of CD4+ blasts spleens were removed from donor mice (C57BL/6) and teased into single-cell suspensions in phosphate-buffered saline (PBS). Erythrocytes were removed by hypotonic lysis. Cells were washed and stimulated with 5 μg/ml ConA for 3 days. CD4+ T blasts were generated by CD8-mediated complement lysis using the αCD8 Ab (53.6-7) and low-toxicity rabbit complement (Biozol, Eching, Germany) followed by positive selection with αCD4-coupled magnetic beads on LS MACS separation columns (Miltenyi Biotec, Germany). Eluted CD4+ T cells were washed and resuspended in sterile PBS, and 3 × 105 purified CD4+ blast was injected into RAG1−/− mice intraperitoneally (i.p.). Cells were tested to be >97% positive for CD4 by flow cytometry. For the second model, CD45RBhigh T cells were purified from spleen cells from donor mice (BALB/c) by two-color sorting on a Moflo cell sorter (Cytomation, Inc., CO, USA). The CD45RBhigh population was defined as the CD4+ cells with the brightest signal for CD45 staining (30% of total CD4+ cells). C.B-17 SCID mice were injected i.p. with 2 × 105 sorted CD45RBhigh CD4+ T cells suspended in 200 μl PBS. The viability of transferred cells was >98% as determined by trypan blue exclusion.
Monoclonal Antibodies
The following monoclonal antibodies (mAbs) were used in vivo: 12-15 (rat IgG1, kindly provided by P. Altevogt, Heidelberg, Germany), directed at the mouse CD2 molecule, and control rat IgG (Sigma, Germany). The following mAbs were used in vitro: 145-2C11 (hamster IgG1, ATCC), directed at murine CD3e; 37.51 (hamster IgG2), directed at murine CD28 (BD Biosciences, Germany); and 53.6-7, directed at murine CD8 (ATCC). The mAb 12-15, 53.6-7, and 145-2C11 were purified from supernatants of the hybridoma cell lines by affinity chromatography employing Protein G Sepharose (Pharmacia, Freiburg, Germany).
mAb Treatment
Treatment with the αCD2 mAb 12-15 was performed in a blinded fashion in four treatment blocks. In transplantation experiments 100 μg of 12-15 per week was found to be as effective as higher concentrations.25 Therefore, mice were loaded with 400 μg followed by 200 μg per week. The treatment blocks consisted of at least four separate blocks each:
-
i)
Prevention of transfer colitis induced by transfer of CD4+ T cell blasts: 400 μg purified 12-15 (n=16) or rat IgG (n=13) was given i.p. just prior to colitis induction, followed by 200 μg weekly thereafter. This treatment block was performed at two different sites (Homburg and Berlin, Germany).
-
ii)
Treatment of established transfer colitis (induced by transfer of CD4+ T-cell blasts) in combination with dexamethasone (1 mg/kg body weight): antibody treatment was started at the beginning of signs of illness (diarrhea or loss of weight > 5%) with 400 μg αCD2 (n=8) or rat-control IgG (n=8) in combination with 1 mg dexamethasone per kg, that is, 15–25 μg per mouse, and was continued with 200 μg antibody combined with dexamethasone (1 mg/kg) on days 1, 4, and subsequently weekly.
-
iii)
Treatment of established transfer colitis induced by transfer of CD4+ T cell blasts (without dexamethasone treatment): treatment began upon first signs of disease (diarrhea or weight loss >5%) with 400 μg 12-15 (n=9) or control–antibody (n=9), followed by weekly administration of 200 μg αCD2 or rat IgG.
-
iv)
Prevention of CD45RBhigh transfer colitis: 400 μg purified 12-15 (n=10) or rat IgG (n=10) was given just prior to colitis induction i.p. followed by 200 μg weekly thereafter.
Mice were monitored for weight loss, rectal prolapse, and diarrhea. Mice that received CD45RBhigh T cells were monitored weekly for hematochezia by hemoccult test from day 21 after transfer. Additionally, 6 weeks after transfer the grade of CD45RBhigh transfer colitis was examined by endoscopy as previously described.35 Blinded videofilms of mouse endoscopy were scored by an experienced endoscopist using a scoring system from 0 to 4 as follows: 0=normal mucosal vascularity and lumen, 1=diminished vascularity, 2=erythema±edema, 3=no vascularity and small ulcers, 4=severe ulceration±stenosis. All mice were killed by cervical dislocation for macroscopical, histological, and cytological examinations when they exhibited one or two of the following signs: weight loss >20%, obvious signs of pain, lethargy.
Blood samples were taken from the heart for blood count analysis using an automatic cytometer (Beckmann Coulter, Germany) and two differential blood smears were prepared. At autopsy spleen, mesenteric lymph nodes (mLN), cecum, and the large intestine were excised. Samples of the cecum and colon were obtained for paraffin sections. The remaining tissues of the large intestine were used for isolation of lamina propria lymphocytes (LPLs).
Depletions Studies
In order to exclude potential effects exerted by the αCD2 mAb 12-15, we transferred 4 × 106 CFSE-labeled CD4+ ConA-blasts to RAG1−/− mice and treated them with either 400 μg 12-15, rat IgG, or GK1.5 (CD4+ depleting antibody) (n=3 for each group). On day 1, we took blood samples and on day 3 recipients were killed. Lymphocytes of peripheral blood cells, spleen, mLN, and lamina propria were isolated as described before. The isolated cells were stained for CD103 (R-PE-conjugated; BD Pharmingen, Heidelberg, Germany) and CD25 (PerCP-CY5.5-conjugated; BD Pharmingen), and CFSE-labeled cells were analyzed by flow cytometry as described below.
CFSE-Labeling, Staining for Intracellular IL-10, and Flow Cytometry
For CFSE-labeling, CD4+ ConA blasts (1 × 107/ml; prepared as described before) were incubated with 1 μM CFSE (10 min, room temperature), washed twice, and transferred to RAG1−/− mice.
At 3 days after transfer, cells were isolated from RAG1−/− recipients (as described above) and stained with saturating amounts of directly conjugated mAb (R-PE-conjugated CD103, Per-CP/anti-CD25-CY5.5-conjugated, BD Pharmingen, Heidelberg) for 10 min at 4°C, washed, and analyzed on the fluorescence-activated cell sorter (FACScan, Becton Dickinson, Heidelberg, Germany).
For intracellular staining of IL-10, isolated cells were stimulated (2 μg/ml ConA, 5 days) and restimulated (10 μg/ml PMA + 1 μg/ml Ionomycin) for 6 h. BrefeldinA (5 μg/ml) was added 3 h before cells were harvested and fixed (2% paraformaldehyde, 10 min). Cells were permeabilized and stained in 0.5% saponin (Merck, Darmstadt) for 10 min at room temperature. Cytokine production was assessed by intracellular staining with FITC-conjugated IL-10 (BD Pharmingen, Heidelberg, Germany) and measured by flow cytometry.
Histological Examination and Microscopic Scoring
Tissue samples of the colon were fixed in 4% formalin and embedded in paraffin. Sections (5 μm) were stained with hematoxylin and eosin for histology. The degree of inflammation was blindly assessed by two investigators using a scoring system, which was modified for the original score as described by Neurath et al36 from 0 to 4 (0, no signs of inflammation; 1, low level of leukocyte infiltration; 2, moderate level of leukocyte infiltration; 3, high level of leukocyte infiltration, high vascular density, and thickening of bowel wall; 4, transmural infiltration, loss of goblet cells, high vascular density, strong bowel wall thickening, ulcerations, and crypt abscesses).
Isolation of Spleen Cells, mLN Cells, and LPL
Single cell suspensions were prepared as described above using a 70 μm mesh cellstrainer (BD Biosciences, Germany). For isolation of lamina propria lymphocytes (LPL), the colon (with rectum) was opened longitudinally and cut into 0.5 cm pieces. The fragments were shaken at 37°C in HBSS (Ca2+/Mg2+-free) containing 1 mM DTT and 2.5 μM EDTA for 45 min with two changes of media. The pellets were resuspended in collagenase medium and shaken for 90 min at 37°C. After digestion of tissue, the suspensions were passed through a 70 μm mesh cellstrainer and centrifuged on a Percoll density gradient (100/40%). LPL were collected from the interphase and resuspended in complete medium (RPMI 1640 medium containing 10% fetal calf serum (FCS), 100 U/mL penicillin/streptomycin, 3 mM glutamine, and 50 μM β-mercaptoethanol). The cell viability was always greater than 90% as determined by trypan blue dye exclusion.
Cytokine Assay
Isolated spleen cells, mLN cells, and LPL (1 × 106 cells/ml in 24-well plates (NUNC, Germany)) were stimulated with coated 145-2C11 (10 μg/ml) and soluble αCD28 mAb 37.51 (1 μg/ml). Supernatants were taken after 48 h and examined for cytokine secretion (IL-2, IL-4, IL-6, IL-10, TNF-α, and IFN-γ) by sandwich ELISA. Antibodies (purified and biotinylated) as well as recombinant protein standards for IL-4, IL-10, and IFN-γ (R&D Systems, Germany) and IL-2, IL-6, and TNF-α (BD Biosciences, Germany) were used according to the manufacturer's instructions.
Proliferation of Spleen Cells, mLNs, and LPL
Isolated cells were incubated in triplicates for 96 h at 5 × 104 cells/well in 96-well round-bottom plates (NUNC, Germany) after stimulation as described above (CD3/CD28). Microcultures received 0.5 μCi of [3H]-thymidine (Amersham Pharmacia, England) per well during the last 18 h of a 96 h culture period and were frozen at −20°C. Incorporated [3H]-thymidine was harvested on glass fiber membranes after rapid thawing of the cultures at 37°C and detected by liquid scintillation counting (LKB Wallace, Finland).
Infection with T. gondii and αCD2 mAb Treatment during Toxoplasmosis
Female C57BL/6 mice were infected with 100 cysts of the ME49 strain of T. gondii by gavage as previously described.34 Overall, 23 mice were blindly treated with either rat IgG (control group, n=8) or the αCD2 mAb 12-15 (n=15) with a loading dose of 200 μg on the day of infection followed by daily doses of 100 μg until day 7. In all, 12 mice were killed on day 12 of infection (four control mice and eight αCD2 mAb-treated mice). The number of parasites per cm ileum was determined as previously described.34 For the remaining 11 mice (four control and seven αCD2 mAb-treated mice), the survival was monitored.
Statistical Analysis
Statistical analysis was carried out using SPSS for Windows. Survival was analyzed using Kaplan–Meier analysis. For other comparisons, the Mann–Whitney U-test was used. Values were expressed as mean (95% confidence intervals) and standard error of mean (s.e.m.). A P value of less than 0.05 was considered significant.
Results
The αCD2 Antibody 12-15 Delays Adoptive Transfer Colitis
To test the preventive potential of the αCD2 mAb 12-15 in transfer colitis, RAG1−/− mice were randomly treated with either αCD2 mAb 12-15 (n=16) or rat IgG (n=13). A starting dose of 400 μg i.p. just prior to CD4 blasts injection was followed by weekly administration of 200 μg i.p. αCD2 mAb treatment significantly prolonged survival using Kaplan–Meier analysis (P<0.001): all rat IgG treated control mice (n=13) died within 35 days, whereas 69% of αCD2 treated animals (n=11) survived beyond 35 days (Figure 1). In all, 50% of the 12-15-treated mice showed no clinical signs of colitis at the end of the experiment, gained weight, and survived until day 75. Histological examinations showed that 12-15-treated mice (n=15) had significantly lower microscopic scores than control IgG treated mice (n=13) (mean±s.e.m.: 2.0±0.3 vs 3.3±0.2, P<0.01). Overall, 12-15-treated mice showed less infiltration, smaller bowel wall thickening as well as less disorganization of the cryptal architecture and regenerating tissue, including mitoses, and eschar formation (Figure 2).

Prolonged survival after preventive treatment of transfer colitis with the αCD2 mAb 12-15. CD4+ ConA blasts were injected into RAG1−/− recipients. Mice initially received 400 μg αCD2 mAb 12-15 (n=16) or rat control IgG (n=13) i.p. and subsequently 200 μg weekly. The αCD2 mAb 12-15 significantly prolongs survival (P<0.001).

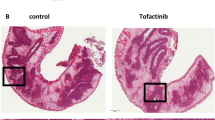
Prevention of adoptive transfer colitis by αCD2 mAb 12-15. The histology of the transverse colon of a RAG1−/− mouse after induction of transfer colitis either following αCD2 mAb (12-15) treatment (a) or rat IgG (b) treatment at weekly intervals starting at 400 μg followed by 200 μg is shown. While αCD2 mAb treatment ameliorates adoptive transfer colitis with a nearly normal histological appearance of the colon (a, × 100), the colon of a rat IgG treated mouse (b, × 100) shows colonic crypt hyperplasia, disorganization, inflammatory infiltrations, strong bowel wall thickening, reduction of goblet cells, as well as crypt abscesses.
When using the classical SCID transfer colitis model (CD45RBhigh CD4+ T cells on SCID mice; n=10 in each group as above) neither αCD2 mAb-treated nor control mice died within 56 days after colitis induction (data not shown). However, only 10% of the αCD2 mAb treated mice had a positive hemoccult test by day 28 as compared to 80% in the control group (Figure 3). Similarly, a significant difference between both groups was found at blinded mouse endoscopy on day 42 (Figure 4). Still, all animals in the αCD2 mAb group eventually developed colitis as jugded by macroscopic and microscopic criteria at the end of the experiment (day 56). Only a trend towards improvement was observed with regard to the macroscopic score (αCD2: 1.5±0.15 vs controls: 1.9±0.22, P<0.1) and no significant difference with regard to the microscopic score (αCD2: 2.3±0.5 vs 2.8±0.4, P=0.3).

αCD2 mAb (12-15) delays development of colitis as assessed by hemoccult tests of the stool. The percentages of positive hemoccult tests in weekly intervals using stool samples of SCID mice that had received 2 × 105 highly purified CD45RBhigh T helper cells on day 0 plus initially 400 μg αCD2 mAb (12-15, n=10) or rat IgG (n=10) i.p. and subsequently 200 μg weekly are shown. *indicates a P<0.05, ** a P<0.005.

αCD2 mAb (12-15) delays development of colitis as assessed by mouse endoscopy. Mouse endoscopy was performed with a rigid endoscope on day 42 after colitis induction as in Figure 3 (SCID transfer model) either in mice receiving αCD2 mAb 12-15 or rat IgG on a weekly basis. (a) the descending colon of a αCD2 mAb 12-15-treated mouse with only slight erythema and normal vascularity and (b) the same colon segment of a rat IgG treated control mouse showing a spontaneously bleeding ulceration and abolished vascularity are shown. (c) Results of 10 mouse colonoscopies are shown as box plot analysis.
Blood count analysis revealed no significant differences with regard to hemoglobin, red cell numbers, and platelet counts in both models (data not shown). However, the αCD2 mAb 12-15 prevented leukocytosis when compared to rat IgG treated control mice (αCD2: 2.97±0.61/μl vs controls: 6.45±0.92/μl , P<0.05) in the T cell blast model, but only a trend was found in the classical SCID transfer colitis model (αCD2: 6.2±1.6/μl vs controls: 8.5.±1.4/μl). Differential blood counts revealed a significant reduction of lymphocytes in both models (T cell blast model: αCD2: 0.23±0.03/μl vs 0.39±0.033/μl, P=0.02; CD45RBhigh model: αCD2: 0.7±0.15/μl vs 2.5±0.44/μl, P<0.001) and a significant reduction of granulocytes only in the T cell blast transfer colitis model (αCD2: 2.74±0.59/μl vs 6.35±1.04/μl, P=0.03). When 4 × 106 CFSE-labeled CD4+ T cell blasts were transferred into RAG1−/− mice, αCD2 mAb treatment did not change total numbers of CFSE-labeled, CD25+/CFSE+, or CD103+/CFSE+ cells neither after 24 nor 72 h in peripheral blood, or at 72 h in mLN, spleen, or LPL when compared to rat IgG treatment (data not shown).
αCD2 Antibody 12-15 Ameliorates Established Colitis
To test the effect of 12-15 in already established colitis we transferred CD4+ T cell blasts into 18 RAG1−/− mice. At the first sign of colitis (diarrhoea and/or weight loss) treatment was started. αCD2 antibody treatment (given on day 0 of established colitis at 400 μg followed by 200 μg weekly) prolonged the average survival after the start of treatment significantly (αCD2 (n=9): 8.6±1.4 days vs rat IgG (n=9): 5.8 ± 0.7; P<0.05) (Figure 5).

αCD2 treatment alone prolongs survival of RAG1−/− mice with established transfer colitis with αCD2. Treatment was started at first signs of colitis. The survival curve after the initiation of treatment is shown. αCD2 treatment (12-15; n=9) or rat control IgG (rat IgG; n=9) was given i.p. initially at 400 μg and subsequently at 200 μg antibody at weekly intervals.
In order to improve the clinical effect of αCD2 mAb treatment, we next asked whether pulse dexamethasone treatment had any additional effect on established colitis. Among 16 transferred RAG1−/− mice, all animals developed colitis within 25 days. αCD2 mAb plus dexamethasone (1 mg/kg) (n=8) or dexamethasone plus rat IgG (control) (n=8) was applied at a mAb/rat IgG dose of 400 μg immediately after colitis was detected (day 0 of established colitis) followed by 200 μg on days 1, 4, and weekly thereafter. When mice with established colitis were treated with αCD2 plus dexamethasone they survived more than twice as long (13.5±2.9 days after initial signs of colitis) as compared with the control group receiving dexamethasone and rat IgG (6.3±1.0 days, P<0.05) (Figure 6). Importantly, dexamethasone did not affect mean survival time when compared to rat IgG alone (24.5 vs 26 days).

Prolonged survival upon αCD2 plus dexamethasone treatment. Mice initially received 400 μg αCD2 (12-15; n=8) or rat control IgG (rat IgG; n=8) i.p., each in combination with dexamethasone (dexa, 1 mg/kg). Subsequent injections were given on day 1, 4, and at weekly intervals (200 μg antibody in combination with 1 mg/kg dexamethasone) thereafter.
αCD2 mAb 12-15 alone or in combination with dexamethasone was not able to decrease clinical, histological or systemic (WBC, RBC, platelets) signs of transfer colitis when given for established colitis.
αCD2 mAb Treatment does not Impair Parasite Control Following Peroral T. gondii Infection
In order to assess potential nonspecific immunosuppression, the influence of αCD2 mAb treatment on infection with T. gondii was studied. Importantly, the number of parasites did not differ in the αCD2 mAb treated vs control group (mean±s.e.m. for αCD2 mice: 334±161 vs control group: 274±69; P=0.7). Furthermore, a trend towards longer survival was found in this model of Th1-mediated immunopathology for αCD2 mAb treated as compared to control mice (P=0.1).
αCD2 mAb Decreases IL-2 Production in CD3/CD28 Stimulated Splenocytes and LPL
Since several studies have shown a Th1 driven immunopathogenesis of transfer colitis, we asked whether αCD2 treatment influences cytokine production. LPL of αCD2 mAb treated animals had a significant reduction of IL-2 production upon stimulation with αCD3/αCD28 mAbs as compared to the control group (n=5 each; preventive experiment; P<0.001; Figure 7a). This finding was confirmed both in splenocytes (P<0.001, Figure 7b, prevention) and mLN cells (P<0.05, Figure 6c, prevention). This was also true for the classical SCID transfer colitis model (data not shown). Treatment with αCD2 in combination with dexamethasone (n=8) led to a significant decrease in IL-2 production in LPL (P<0.05), and to a slight but nonsignificant decrease in splenocytes (P<0.1) and mLN cells (P<0.1). αCD2 alone was not able to decrease IL-2 production in LPL, spleen, or mLN cells in established colitis (data not shown).

αCD2 mAb inhibits IL-2 production. LPL (a), spleen (b), and mLN cells (c) were isolated from colitic RAG1−/− mice treated either with αCD2 mAb (12-15) or control IgG starting at transfer (prevention). Established colitis was treated with αCD2 mAb (12-15) or rat control IgG plus 1 mg/kg dexamethasone at days 0, 1, 4, 7, 14 after first signs of colitis. Cells were isolated from the colon of colitic mice and activated for two days with αCD3 and αCD28 mAb. IL-2 was measured by ELISA and is expressed as box plots. The level of significance is indicated (*P<0.05; ***P<0.001) and was determined by the Mann–Whitney U-test.
No differences were found with regard to production of the cytokines IL-6, IL-10, IFN-γ, and TNF-α in all three experiments using the T cell blast transfer model. In contrast, TNF-α levels in the classical SCID transfer colitis model significantly decreased in the supernatant of LPLs but not splenocytes or mLN. IL-4 was not increased in the αCD2 mAb treated groups as compared to the rat IgG treated control groups (data not shown). Importantly, no differences were seen when looking at intracellular IL-10 levels by flow cytometry (data not shown).
αCD2 Decreases Proliferation of CD3/CD28 Stimulated Cells
To test whether αCD2 could alter other T cell functions, proliferation of splenocytes, mLN cells, and LPL was investigated. Spleen and mLN cells from αCD2 mAb-treated animals had a significantly decreased proliferative response to CD3/CD28 stimulation in vitro (Figure 8a, splenocytes of preventively treated mice (P<0.01), Figure 8b, mLN cells of preventively treated mice (P<0.05)). Overall proliferation of LPL was too low for comparison (not shown).

Significant inhibition of CD3/CD28 stimulated T cell proliferation after treatment with αCD2 mAb 12-15. (a) Splenic lymphocytes and (b) mLN cells from colitic RAG1−/− mice treated preventively with 12-15 or rat-control IgG, respectively, are shown. Established colitis was treated with αCD2 mAb 12-15 or rat IgG (initially 400 μg followed by 200 μg, days 0, 1, 4, 7, 14) in combination with 1 mg/kg dexamethasone once colitis was clinically overt. After in vitro stimulation with αCD3-coated plates and soluble αCD28, cells were cultured and proliferation was determined by [3H]-thymidine uptake. The proliferative indices (counts of sample/baseline counts) are expressed as box plots. The level of significance is indicated (*P<0.05; **P<0.01) and was determined by the Mann–Whitney U-test.
αCD2 mAb in combination with dexamethasone also reduced the proliferative response when compared to dexamethasone plus control rat IgG; however, this difference was only significant in mLN cells, not in spleen cells (Figure 8a, established colitis: αCD2+dexa, splenocytes, Figure 8b, established colitis: αCD2+dexa, mLN cells (P<0.05)).
Mice treated with αCD2 mAb alone showed no decrease in proliferation of stimulated splenocytes, mLN cells, or LPL.
Discussion
In this study, we demonstrate for the first time the ability of an αCD2 mAb to delay the development of adoptive transfer colitis and to decrease the signs of inflammation in established transfer colitis. Most likely, this beneficial effect results from markedly reduced IL-2 production and inhibition of T-lymphocyte proliferation, two characteristics of adoptive transfer colitis. Presumably, due to the severity of the model, this immunotherapy neither prevents colitis completely nor does it cure established disease. Still, αCD2 treatment effects are almost as good as previous reports on αTNF–α mAb treatment in CD45RBhigh transfer colitis,37 a therapy that turned out to be effective in refractory Crohn's disease.38
An important histological feature of inflammatory bowel disease is the strong mononuclear cell infiltrate consisting mainly of CD4+ T lymphocytes. Increased proliferation,1 defective apoptosis,2, 3 increased adhesion,39 and enhanced lymphokine production1, 40 are generally thought to be responsible. In order to investigate these processes and to obtain an effective immunotherapy for IBD, animal models are a useful tool. The most widely used model is the adoptive transfer colitis having a Th1 dominance, which exists with many variations mainly using either sorted CD45RBhigh T helper cells or alternatively T cell blasts.41, 42 Enhanced adhesion as well as increased colonic T cell proliferation and apoptosis are additional features of this model.43, 44 Therefore, adoptive transfer colitis serves as a useful model to investigate the role of T cells in IBD, particularly with regard to cytokine dysbalance (Th1 dominance as in Crohn's disease), increased proliferation, and probably enhanced adhesion. In contrast to other IBD models the colitis induced by transfer of CD4+ T cell blasts to SCID mice or RAG1−/− mice results in highly reproducible severe colitis with 100% mortality within 35 days. Our results show that indeed mAb directed at the T cell antigen CD2 have a positive effect on survival and the degree of inflammation in transfer colitis. Additional experiments using the classical CD45RBhigh transfer colitis model revealed that the αCD2 mAb delays but does not prevent colitis development as seen with delayed hematochezia and reduced mucosal inflammation when using endoscopy on day 42.
Previous studies have shown that αCD2 mAbs can ameliorate chronic inflammatory diseases, that is, adjuvant arthritis,29 or prolong transplantation survival in different species.25, 26, 27, 28 So far, αCD2 mAb treatment has only been tested in one IBD animal model, that is, rat 2,4,6-trinitrobenzene sulphonic acid-induced colitis.45 As this model turned out to be T cell independent, we were unable to find a beneficial effect by αCD2 mAb in this rat colitis model. In contrast, we now show that CD2-directed immunotherapy can partially prevent T cell-mediated colitis and can attenuate already established colitis, particularly in combination with dexamethasone.
The most likely underlying mechanism of the αCD2 mAb treatment is its inhibitory effect on mLN/splenocyte proliferation as well as reduced IL-2 production by LPL, mLN, and splenocytes, suggesting that IL-2 serves as an autocrine growth factor. This previously reported46 and highly reproducible effect probably leads to a reduction of absolute lymphocyte numbers at the end of the experiments. Since mitogenic αCD2 mAb can strongly induce IL-2 production via the AP-1 signalling pathway,47 inhibition of IL-2 production is probably due to the sterical blockade of the CD2 molecule. In addition, we were unable to find evidence of the induction of regulatory T cells as measured by flow cytometry of CD25 and CD103 expression of the transferred T cells. This again confirms previous reports that the αCD2 mAb 12-15 has no effect on CD25 expression.48 Further, no effect was seen with regard to IL-10 secretion by splenocytes, mLN, or LPL upon CD3/CD28 stimulation. Therefore, it seems highly unlikely that this particular αCD2 mAb induces classical regulatory T cells. This seems to be important, because human studies suggest that regulatory T cells are induced via CD2.18 Finally, in our hands this mAb did not induce Th2 cytokines as reported by Chavin et al.22
Apart from inhibition of proliferation20, 21, 49 and a reduction of IL-2 production50 αCD2 mAb have previously been shown to induce T cell apoptosis of LPL16 and to block T cell adhesion leading to reduced tumor cytotoxicity.51 In the past, we found that apoptosis induction via human CD2 usually only occurs when proliferation is induced at the same time (Hoffmann et al, submitted). Others were not able to find peripheral T cell depletion by this αCD2 mAb,48 while we found a moderate reduction of lymphocytes probably due to less T cell proliferation rather than apoptosis induction.
Originally, CD2 was discovered via its role in in vitro and later in vivo tumor cytotoxicity.20, 51 However, studies using CD2 deficient mice did not show an increased tumor incidence or general immunsuppression as demonstrated by normal cellular immune responses upon infection with lymphocytic choriomeningitis virus or Pneumocystis carinii.52, 53 Treatment of Crohn's disease patients and patients with rheumatoid arthritis with αTNF α mAb recently taught us that specific immunotherapies can not only potently suppress disease activity but also reactivate latent infections such as tuberculosis, sometimes with fatal consequences.33 Previous studies have shown that Th1 cells (ie secreting TNF-α, interferon γ and NO) are required for parasite control in the murine model of oral infection with T. gondii.34, 54 Importantly, the αCD2 mAb described here did not affect parasite control. Therefore, there is no evidence that the CD2-directed immunotherapy leads to significant immunosuppresion in spite of reducing lymphocyte counts, of inhibition of T cell proliferation, and/or of reduction of IL-2 production.
In spite of the prolonged survival time of most transfer colitis mice, the αCD2 mAb did not prevent colitis in all mice, and its clinical efficacy in established colitis is limited. In addition, all SCID mice that received CD45RBhigh T helper cells eventually developed colitis when looking at a later time point. This finding supports previous transplantation studies suggesting that this αCD2 mAb prolongs allogenic heart transplantion.25 However, tolerance induction can only be achieved upon combination with αCD3, CTLA-4-Ig, or αCD48 mAb.55, 56, 57 One way of enhancing clinical effectiveness in our study was to add pulse dexamethasone, which did not alter the disease course on its own. The theoretical benefit of higher doses of αCD2 mAb or shorter injection intervals stands against previous transplantation studies using much lower doses of the same mAb.25
In conclusion, we have described amelioration or delay of adoptive transfer colitis and attenuation of established colitis by treatment with the αCD2 mAb 12-15. The data demonstrate that a blocking αCD2 mAb can profoundly alter the immune response in a murine model of intestinal inflammation induced by transfer of CD4+ blasts into immunodeficient RAG1−/− mice or CD45RBhigh T cells into SCID mice probably by inhibiting proliferation and decreasing IL-2 production. Still, αCD2 mAb does not impair parasite control in a Toxoplasmosis model. Future studies have to investigate how a CD2 directed immunotherapy can be optimized in IBD animal models and how these findings can be applied to clinical CD2-directed immunotherapies.
References
Autschbach F, Schürmann G, Qiao L, et al. Cytokine messenger RNA expression and proliferation status of intestinal mononuclear cells in noninflamed gut and Crohn's disease. Virchows Arch 1995;426:51–60.
Boirivant M, Marini M, Di Felice G, et al. Lamina propria T cells in Crohn's disease and other gastrointestinal inflammation show defective CD2 pathway-induced apoptosis. Gastroenterology 1999;116:557–565.
Ina K, Itoh J, Fukushima K, et al. Resistance of Crohn's disease T cells to multiple apoptotic signals is associated with a Bcl-2/Bax mucosal imbalance. J Immunol 1999;163:1081–1090.
Sans M, Panes J, Ardite E, et al. VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology 1999;116:874–883.
Farkas S, Herfarth H, Rossle M, et al. Quantification of mucosal leucocyte endothelial cell interaction by in vivo fluorescence microscopy in experimental colitis in mice. Clin Exp Immunol 2001;126:250–258.
Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol 1996;157:1261–1270.
Kakazu T, Hara J, Matsumoto T, et al. Type 1 T-helper cell predominance in granulomas of Crohn's disease. Am J Gastroenterol 1999;94:2149–2155.
Strober W, Fuss IJ, Blumberg RS . The immunology of mucosal models of inflammation. Annu Rev Immunol 2002;20:495–549.
van der Merwe PA . Formation and function of the immunological synapse. Curr Opin Immunol 2002; 14:293–298.
van der Merwe PA, Davis SJ . Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol 2003;21:659–684.
Shaw S, Luce GE, Quinones R, et al. Two antigen-independent adhesion pathways used by human cytotoxic T-cell clones. Nature 1986;323:262–264.
Bierer BE, Peterson A, Gorga JC, et al. Synergistic T cell activation via the physiological ligands for CD2 and the T cell receptor. J Exp Med 1988;168:1145–1156.
Pirzer UC, Schürmann G, Post S, et al. Differential responsiveness to CD3-Ti vs. CD2-dependent activation of human intestinal T lymphocytes. Eur J Immunol 1990;20:2339–2342.
Zeitz M, Quinn TC, Graeff AS, et al. Mucosal T cells provide helper function but do not proliferate when stimulated by specific antigen in lymphogranuloma venereum proctitis in nonhuman primates. Gastroenterology 1988;94:353–366.
Targan SR, Deem RL, Liu M, et al. Definition of a lamina propria T cell responsive state. Enhanced cytokine responsiveness of T cells stimulated through the CD2 pathway. J Immunol 1995;154:664–675.
Boirivant M, Pica R, DeMaria R, et al. Stimulated human lamina propria T cells manifest enhanced Fas-mediated apoptosis. J Clin Invest 1996;98:2616–2622.
Itoh J, Kugathasan T, McCormick T, et al. Resistence of Crohn's disease (CD) mucosal T-cells to cell death is independent of trigger: Decreased anti-Fas and nitric oxide (NO)-induced apoptosis. Gastroenterology 1998; 114:G1553.
Wakkach A, Cottrez F, Groux H . Differentiation of regulatory T cells 1 is induced by CD2 costimulation. J Immunol 2001;167:3107–3113.
Bullens DM, Rafiq K, Charitidou L, et al. Effects of co-stimulation by CD58 on human T cell cytokine production: a selective cytokine pattern with induction of high IL-10 production. Int Immunol 2001;13:181–191.
Krensky AM, Sanchez-Madrid F, Robbins E, et al. The functional significance, distribution, and structure of LFA-1, LFA-2, and LFA-3: cell surface antigens associated with CTL- target interactions. J Immunol 1983;131:611–616.
Palacios R, Martinez Maza O . Is the E receptor on human T lymphocytes a ‘negative signal receptor’? J Immunol 1982;129:2479–2485.
Chavin KD, Qin L, Yon R, et al. Anti-CD2 mAbs suppress cytotoxic lymphocyte activity by the generation of Th2 suppressor cells and receptor blockade. J Immunol 1994;152:3729–3739.
Peng X, Kasran A, Bullens D, et al. Ligation of CD2 provides a strong helper signal for the production of the type 2 cytokines interleukin-4 and -5 by memory T cells. Cell Immunol 1997;181:76–85.
Gray JD, Hirokawa M, Ohtsuka K, et al. Generation of an inhibitory circuit involving CD8+ T cells, IL-2, and NK cell-derived TGF-beta: contrasting effeccts of anti-CD2 and anti-CD3. J Immunol 1998;160:2248–2254.
Chavin KD, Lau HT, Bromberg JS . Prolongation of allograft and xenograft survival in mice by anti- CD2 monoclonal antibodies. Transplantation 1992;54:286–291.
Przepiorka D, Phillips GL, Ratanatharathorn V, et al. A phase II study of BTI-322, a monoclonal anti-CD2 antibody, for treatment of steroid-resistant acute graft-versus-host disease. Blood 1998;92:4066–4071.
Mourad M, Besse T, Malaise J, et al. BTI-322 for acute rejection after renal transplantation. Transplant Proc 1997;29:2353.
Haga M, Hirahara H, Tsuchida M, et al. Costimulatory signal blockade by anti-CD2 monoclonal antibody in combination with 15-deoxyspergualin prolongs concordant xenograft survival. Transplant Proc 2000;32:1006–1008.
Hoffmann JC, Herklotz C, Zeidler H, et al. Anti-CD2 (OX34) MoAb treatment of adjuvant arthritic rats: attenuation of established arthritis, selective depletion of CD4+ T cells, and CD2 down-modulation. Clin Exp Immunol 1997;110:63–71.
Hoffmann JC, Herklotz C, Zeidler H, et al. Initiation and perpetuation of rat adjuvant arthritis is inhibited by the anti-CD2 monoclonal antibody (mAb) OX34. Ann Rheum Dis 1997;56:716–722.
Jung S, Toyka K, Hartung HP . Suppression of experimental autoimmune encephalomyelitis in Lewis rats by antibodies against CD2. Eur J Immunol 1995;25:1391–1398.
Ellis CN, Krueger GG . Treatment of chronic plaque psoriasis by selective targeting of memory effector T lymphocytes. N Engl J Med 2001;345:248–255.
Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha- neutralizing agent. N Engl J Med 2001;345:1098–1104.
Liesenfeld O, Kosek J, Remington JS, et al. Association of CD4+ T cell-dependent, interferon-gamma-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J Exp Med 1996;184:597–607.
Becker C, Fantini MC, Schramm C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004;21:491–501.
Neurath MF, Fuss I, Kelsall BL, et al. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med 1995;182:1281–1290.
Powrie F, Leach MW, Mauze S, et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1994;1:553–562.
Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med 1997;337:1029–1035.
Van Assche G, Rutgeerts P . Antiadhesion molecule therapy in inflammatory bowel disease. Inflamm Bowel Dis 2002;8:291–300.
Mullin GE, Lazenby AJ, Harris ML, et al. Increased interleukin-2 messenger RNA in the intestinal mucosal lesions of Crohn's disease but not ulcerative colitis. Gastroenterology 1992;102:1620–1627.
Powrie F, Leach MW, Mauze S, et al. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol 1993;5:1461–1471.
Rudolphi A, Boll G, Poulsen SS, et al. Gut-homing CD4+ T cell receptor alpha beta+ T cells in the pathogenesis of murine inflammatory bowel disease. Eur J Immunol 1994;24:2803–2812.
Bregenholt S, Reimann J, Claesson MH . Proliferation and apoptosis of lamina propria CD4+ T cells from scid mice with inflammatory bowel disease. Eur J Immunol 1998;28:3655–3663.
Burns RC, Rivera-Nieves J, Moskaluk CA, et al. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn's disease in mice. Gastroenterology 2001;121:1428–1436.
Hoffmann JC, Peters K, Henschke S, et al. Role of T lymphocytes in rat 2,4,6-trinitrobenzene sulphonic acid (TNBS) induced colitis: increased mortality after gammadelta T cell depletion and no effect of alphabeta T cell depletion. Gut 2001;48:489–495.
Chavin KD, Qin L, Lin J, et al. Combination anti-CD2 and anti-CD3 monoclonal antibodies induce tolerance while altering interleukin-2, interleukin-4, tumor necrosis factor, and transforming growth factor-beta production. Ann Surg 1993;218:492–501.
Gonsky R, Deem RL, Hughes CC, et al. Activation of the CD2 pathway in lamina propria T cells up-regulates functionally active AP-1 binding to the IL-2 promoter, resulting in messenger RNA transcription and IL-2 secretion. J Immunol 1998;160:4914–4922.
Lin J, Yon RW, Chavin KD, et al. Anti-CD2 monoclonal antibody-induced receptor changes: down modulation of cell surface CD2. Transplantation 1995;59:1162–1171.
Chavin KD, Qin L, Lin J, et al. Anti-CD2 and anti-CD3 monoclonal antibodies synergize to prolong allograft survival with decreased side effects. Transplantation 1993;55:901–908.
Reed JC, Tadmori W, Kamoun M, et al. Suppression of interleukin 2 receptor acquisition by monoclonal antibodies recognizing the 50 KD protein associated with the sheep erythrocyte receptor on human T lymphocytes. J Immunol 1985;134:1631–1639.
Gückel B, Berek C, Lutz M, et al. Anti-CD2 antibodies induce T cell unresponsiveness in vivo. J Exp Med 1991;174:957–967.
Evans CF, Rall GF, Killeen N, et al. CD2-deficient mice generate virus-specific cytotoxic T lymphocytes upon infection with lymphocytic choriomeningitis virus. J Immunol 1993;151:6259–6264.
Beck JM, Blackmon MB, Rose CM, et al. T cell costimulatory molecule function determines susceptibility to infection with Pneumocystis carinii in mice. J Immunol 2003;171:1969–1977.
Liesenfeld O, Kang H, Park D, et al. TNF-alpha, nitric oxide and IFN-gamma are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite Immunol 1999;21:365–376.
Chavin KD, Qin L, Lin J, et al. Combined anti-CD2 and anti-CD3 receptor monoclonal antibodies induce donor-specific tolerance in a cardiac transplant model. J Immunol 1993;151:7249–7259.
Woodward JE, Qin L, Chavin KD, et al. Blockade of multiple costimulatory receptors induces hyporesponsiveness: inhibition of CD2 plus CD28 pathways. Transplantation 1996;62:1011–1018.
Qin L, Chavin KD, Lin J, et al. Anti-CD2 receptor and anti-CD2 ligand (CD48) antibodies synergize to prolong allograft survival. J Exp Med 1994;179:341–346.
Acknowledgements
The work of the authors was supported by DFG Grant No1561/3-5, SFB 633/B3, SFB 633/B6, and German Competence Network IBD (BMBF/DLR). We thank P Altevogt, German Cancer Research Center, Heidelberg, Germany, for providing us with the αCD2 hybridoma 12-15.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pawlowski, N., Kakirman, H., Kühl, A. et al. αCD2 mAb treatment safely attenuates adoptive transfer colitis. Lab Invest 85, 1013–1023 (2005). https://doi.org/10.1038/labinvest.3700295
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700295
Keywords
This article is cited by
-
γδ T lymphocytes: a new type of regulatory T cells suppressing murine 2,4,6-trinitrobenzene sulphonic acid (TNBS)-induced colitis
International Journal of Colorectal Disease (2008)
