
Abstract
Here we review mechanisms and molecules that necessitate protection and oppose axonal growth in the injured spinal cord, representing not only a cast of villains but also a company of therapeutic targets, many of which have yet to be fully exploited. We next discuss recent progress in the fields of bridging, overcoming conduction block and rehabilitation after spinal cord injury (SCI), where several treatments in each category have entered the spotlight, and some are being tested clinically. Finally, studies that combine treatments targeting different aspects of SCI are reviewed. Although experiments applying some treatments in combination have been completed, auditions for each part in the much-sought combination therapy are ongoing, and performers must demonstrate robust anatomical regeneration and/or significant return of function in animal models before being considered for a lead role.
Similar content being viewed by others

Introduction
For clinician and scientist alike, the injured spinal cord is a formidable scene: a large cast of endogenous cells and molecules act in concert to prevent or restrict functional connectivity. The players and their roles in limiting spinal cord repair continue to be elucidated, and each discovery unveils the character of a molecular antagonist that hinders return of function after spinal cord injury (SCI). As the villains reveal themselves, our repertoire of possible targets for intervention grows, and potential treatments fall into one or more of five categories: (1) protection, to prevent death of neural cells undamaged by the initial injury; (2) stimulating axonal growth, either by enhancing the intrinsic regenerative capacity of spinal and supraspinal neurons or by blocking or removing endogenous inhibitors to repair; (3) bridging, to provide a permissive substrate for elongating axons and to replace lost tissue; (4) enhancing axonal transmission, to alleviate conduction block in spared or regenerated axons and (5) rehabilitation, to enhance functional plasticity within surviving circuits and consolidate anatomical repair.
As the cast of villains in SCI is vast and collaborative, so too must be the chorus of heroes that rise to meet them. It is widely acknowledged that a combination of treatments will be required to address the complex issues of SCI;1 however, designing such a treatment strategy is incredibly daunting. The first step is to identify the most powerful and/or versatile performers to fill the roles of protection, stimulating growth, bridging, conduction and rehabilitation: each treatment must audition in isolation before it can take the stage as part of a combination therapy. The second step is equally ambitious: once the protagonists of SCI are identified, they must appear in the appropriate spatial and temporal combination. Like a performance, the development of a reliable therapy for SCI must be directed with intimate knowledge of each character, leading to the perfect balance among the players.
Neuroprotection in the setting of SCI
Although traumatic SCI is often the result of a single mechanical insult, the functional outcome is determined by a cascade of behind-the-scenes events described collectively as ‘secondary injury’. Displacement or penetration of the spinal cord both inflicts direct tissue damage (ie primary injury) and initiates destructive processes that expand the injury site. This phenomenon of secondary injury, which extends for days after the initial trauma, presents the first opportunity for intervention after SCI. Events that fall under the umbrella of secondary injury include vascular changes (edema, ischemia, and hypoxia), excitotoxic biochemical events (formation of free radicals and nitric oxide, protease release), and cellular responses (invasion by immune cells, activation of glial cells, and death of neurons and glia) (reviewed in Hausmann2). How significantly each component of secondary injury contributes to loss of function remains unclear: what is evident is that the resulting pathology might be ameliorated by eliminating one or more of these players in the early stages of SCI.
Clinical trials of neuroprotective agents, delivered acutely following SCI, have been carried out with equivocal success. The two most widely discussed are the National Acute Spinal Cord Injury Study (NASCIS) trial of high-dose systemic methylprednisolone (MP)3 and the Sygen® trial of GM-1 gangliosides.4 GM-1 gangliosides appeared to enhance the rate of recovery from SCI, but did not significantly improve the ultimate functional outcome, whereas MP was reported to improve functional outcome when administered within 8 h of SCI.5 Although MP has since been widely used as a neuroprotective therapy, several more recent studies have reported equivocal benefits and concerns about the potential toxicity of the required high doses and observed medical complications (reviewed by Kwon et al6). Administration of this steroid is no longer a standard treatment, nor even a guideline for treatment, of acute SCI in Canada7 and many other countries have adopted similar policies.
In the midst of controversy surrounding the use of currently available pharmacologic interventions, the search for an effective neuroprotective agent is ongoing in animal models of SCI. Neuroprotective strategies have been reviewed at length previously.8, 9 Here we focus on recent work from our group and elsewhere dealing with some of the neuronal consequences of secondary injury, including axonal retraction/dieback and neuronal atrophy/death.
One potential mechanism for secondary neuronal injury which is undergoing thorough investigation in a number of laboratories involves endogenous (microglial) or invading (hematogenous) phagocytic cells. While the potential therapeutic use of previously myelin-activated macrophages has received much attention of late (reviewed in;10 see ‘Myelin and myelin signaling’, below), several lines of evidence demonstrate that the microglial/macrophage response to SCI is responsible for demyelination, oligodendrocyte death, and damage to intact axons.11, 12, 13, 14 The nontraumatic injection of the micgroglia/macrophage activator zymosan into the brain and spinal cord results in cavitation, demyelination, and axonal injury.15, 16 The molecules released by mature phagocytes, including superoxide, hydrogen peroxide, hypochlorous acid, as well as neurotoxic chemokines and cytokines may be partially responsible for axonal damage and retraction, and neuronal death.17
The second-generation tetracycline derivative, minocycline, has been shown to be an antiinflammatory agent. Following SCI, systemic minocycline administration has also been documented to inhibit microglial activation, to promote oligodendrocyte survival, to reduce lesion-induced cavity formation, and to prevent the retraction or dieback of injured dorsal column, rubrospinal, and corticospinal axons.18, 19, 20 The axonal protection afforded by this treatment may be of interest, since the rats also exhibit improved functional recovery,18, 19, 21 although the restricted subtotal nature of the dorsal column lesion would permit neural plasticity within preserved pathways to contribute to recovery. The mechanism by which sensory dorsal column axons dieback following injury may involve macrophage-mediated degeneration of proximal myelin sheaths, since in myelin-deficient rats macrophage/microglial activation with zymosan does not induce axonal retraction.20 Interestingly, suppression of p75, a putative signaling receptor for myelin inhibition, also reduced axonal retraction/dieback from the lesion site in p75 knockout mice (p75−/−).22 This effect of p75 deletion was evident in pictures of the lesion site, although the authors of the study did not discuss reduced retraction/dieback as a result of this study.
The loss of neurons and glia at the site of SCI is well-documented. However, at more remote distances, the extent of neuronal death following SCI has probably been overestimated. Even 1 year after a cervical SCI, the cell bodies of severed descending rubrospinal axons, which were previously thought to have perished, regained normal neuronal morphology when BDNF was administered in the vicinity of the red nucleus.23 Rather than being generated de novo, these rubrospinal neurons had atrophied to the point where they were easily missed during previous histological screens, leading to an underestimation of their numbers and resulting in the premature declaration of their death. Recently, adeno-associated viral (AAV) vector-mediated BDNF gene transfer into the red nucleus similarly counteracted atrophy of chronically lesioned rubrospinal neurons.24 It is not known to what extent severe atrophy (rather than neuronal loss) is a general response of CNS neurons to chronic injury, but corticospinal neurons also exhibit atrophy after spinal axotomy.25 If SCI-induced atrophy is common among CNS neurons, it may mean that rather than attempting to replace neurons through transplantation of embryonic tissue or multipotent cells,26 therapies directed at reviving these ‘hibernating’ neurons may be more appropriate. It has been observed that the direct CNS application of neurotrophic factors following SCI can prevent the atrophy of rubrospinal,27, 28 corticospinal,25, 29 and ascending propriospinal neurons,30 as well as stimulate the expression of axonal growth-associated genes. Many of the same factors can also prevent corticospinal and ascending sensory axonal dieback.31
Stimulating axonal growth in the injured spinal cord
At the site of SCI, axons attempting to regrow and/or re-establish functional connections get a chilly reception, not only because of necrotic and apopotic forms of secondary cell death and the activation of immune cells (discussed in the previous section above), but also because a troupe of inhibitory molecules confront growth cones at and around the site of SCI. These molecules can be broadly classified into two groups: myelin-associated molecules produced by oligodendrocytes, and extracellular matrix (ECM) molecules, many of which are produced by astrocytes in response to injury. Nerve grafting experiments and other manipulations (discussed in the section on Bridging the site of SCI below) indicate that damaged CNS axons can regenerate when their local environment is replaced with a substrate that is less inhibitory (or more permissive). Even so, it has been demonstrated repeatedly that the capacity for axonal regeneration is hindered by an impoverished neuronal growth program being generated by severed neurons. Cajal was perhaps the first to recognize that ‘…for these [growth cones] to become consolidated and to progress, the action of the trophic center or nerve cell is indispensable from every point of view.’32 In this section, we will consider both the molecular antagonists to regeneration after SCI, and the protagonistic manipulations that might coax CNS axons into a state of growth in their native environment, through direct targeting of the responsible inhibitory factors within CNS tissue, the axonal responses to those inhibitors, or through enhancing the response of the cell body to injury.
Myelin and myelin signaling: an inhibitory chorus line
In the normal CNS, myelin plays a critical role in solidifying connections established in development. After SCI, myelin-derived molecules take the stage in a new light, as critical inhibitors of axonal growth/regeneration. The role of myelin in suppressing plasticity after SCI was first illustrated in landmark experiments employing the Nogo-blocking antibody (IN-1):33, 34 antagonizing Nogo, the now notorious inhibitory constituent of central myelin, permitted extensive growth of cut corticospinal axons in young rats. In fact, Nogo – now known to exist in three isoforms35, 36 – is but one of a number of myelin-derived inhibitory proteins. Others identified thus far include myelin-associated glycoprotein (MAG)37, 38 and oligodendrocyte myelin glycoprotein (OMgp).39
The role of MAG as an impediment to axonal regeneration is exemplified by in vitro studies in which its immunodepletion from myelin extracts attenuated inhibitory activity.37 It has since been shown that elevating cAMP can stimulate adult neurons to extend axons on MAG substrates,40 an effect related to the growth-enhancing effects of conditioning lesions and neurotrophic factor application (see below). Studies in MAG knockout mice have been equivocal, however, since myelin preparations from these mice are still effective in blocking regeneration, and since severed optic nerve and corticospinal tract (CST) axons regenerate no better in MAG knockouts than in wild-type mice.41 Recent characterization of plasticity following SCI in three lines of mice lacking one or more Nogo molecules has been similarly perplexing. While corticospinal axon sprouting following spinal hemisection was evident and apparently robust in some mice,42, 43 almost no augmentation of regeneration or sprouting was seen in others.44 Immunizing mice with MAG-plus-NogoA enhances regeneration and/or sprouting of corticospinal axons following spinal hemisection; however, myelin immunization has a greater overall effect than MAG/NogoA immunization.45, 46 Manipulations of OMgp have yet to be carried out, but one might hypothesize that, like the focused targeting of MAG or Nogo alone, specifically reducing OMgp activity would have less effect on axonal regeneration than targeting multiple myelin-derived inhibitors.
Fortuitously, recent data indicate that this trio of myelin-derived inhibitors – NogoA, MAG and OMgp – signal through a common receptor (the Nogo receptor, NgR).47, 48, 49, 50 As an apparent point of convergence in inhibitory signaling, NgR has taken the stage as a target of strategies aimed at increasing plasticity in the injured spinal cord. A competitive antagonist of NgR, NEP1-40, blocks the inhibitory action of myelin in vitro51 and enhances both growth of supraspinal axons and functional recovery when administered intrathecally or systemically after thoracic hemisection.51, 52 Another antagonist, NgREcto, is a soluble, truncated form of NgR, which has been shown to alleviate inhibition of both Nogo and myelin in vitro (Fournier et al, 2002). Recent work from our group examines the effects of NgR activity using a different form of soluble NgR in vivo;53 in these experiments, we found that NgR antagonism augmented rhizotomy-induced sprouting of both descending monoaminergic axons and peptidgeric primary afferents in the cervical dorsal horn. The discovery of an anti-NgR monoclonal antibody, capable of inhibiting binding of Nogo, MAG, and OMgp, and of blocking inhibition of myelin in vitro, has now been reported.54
Downstream of NgR are several other molecules that might also serve as targets for intervention after SCI. The Nogo receptor is thought to transmit myelin-inhibitory signaling through its interaction with the low-affinity neurotrophin receptor p75.55, 56 Recently, LINGO-1, a neural transmembrane protein, has been revealed as an essential component of the functional NgR/p75 complex.57 Ligand-bound NgR associates with p75 and LINGO-1 to activate the GTPase RhoA, which transduces myelin-derived inhibition to the cytoskeleton.58, 59, 60 Since p75 binds neurotrophins, its contribution to axonal growth is complex (see below), whereas the activation of RhoA leads to the recruitment of Rho kinase and subsequent remodeling of the actin cytoskeleton to inhibit regeneration. Targeting the Rho pathway with the bacterial enzyme C3 transferase or with specific inhibitors of Rho kinase has led to reports of both corticospinal regeneration and functional recovery after rat SCI.61, 62 Interestingly, it seems that astrocyte or meningeal cell-derived ECM molecules may also signal through the Rho pathway,63, 64, 65 indicating a convergence of multiple inhibitory signaling pathways, as well as suggesting that multiple tiers of inhibition can be overcome by focusing on a single intracellular pathway (for a review, see Kwon et al66).
It is likely that other myelin-associated inhibitors remain to be elucidated. A broader, approach, then, is to temporarily and focally remove myelin and/or myelin debris from the site of SCI. We have used a complement-fixing antibody to galactocerebroside (GalC), coadministered intraspinally with serum complement, to achieve transient focal demyelination at the site of SCI.67, 68, 69 Within the CNS, GalC is restricted to the cell membrane of oligodendrocytes and myelin; specifically, it is present in the outer layer of the myelin membrane. Binding of the GalC antibody triggers activation of the complement cascade, which results in disruption of myelin membranes and recruitment of endogenous microglia or invading macrophages to phagocytically clear CNS myelin within the injured region. Using this approach, the attack on myelin is volume- and concentration-dependent, can be readily tracked with noninvasive magnetic resonance imaging, ceases when the infusion is stopped, and is reversible, as remyelination occurs when treatment is withdrawn. Myelin disruption with the GalC antibody permits regeneration of brainstem-spinal axons when administered after acute hemisection in the adult rat.68 More recently, we have found that immunological myelin disruption stimulates both regeneration of supraspinal axons and improved recovery of locomotion after severe thoracic contusion injuries (where controls are permanently paralyzed), even when treatment is delayed for 1 or 2 months after injury (JDS, unpublished observations).
It has been suggested that another method of reducing myelin-derived inhibition at the site of SCI is to transplant autologous macrophages at the lesion site to phagocytically clear myelin debris. In rat, macrophages activated by prior exposure to degenerating peripheral nerve segments stimulated both regeneration and functional recovery when grafted at the site of complete thoracic transection;70 more recently, macrophages coincubated with skin improved functional outcome when grafted at the site of severe thoracic contusion.71 Activated autologous macrophages have entered clinical trials as ProCord, a therapy administered by ProNeuron, based in Israel. Although patients have already received ProCord, no published reports of their progress are available. As all patients received ProCord within 14 days of SCI, the effect of stimulated macrophage transplantation on their clinical outcome may prove difficult to interpret: in spite of this potential limitation, a Phase II multicenter trial has been initiated (http://www.proneuron.com/ClinicalStudies/index.html). Despite their foray into clinical testing, macrophage transplantation remains a controversial therapy for SCI, largely because the activation of macrophages after SCI may also have other actions within the CNS, which have yet to be characterized.72
Astrocytes and the glial scar: another inhibitory ensemble
An excellent setting in which to study the astroglial contributions to inhibition of regeneration is that of the dorsal root entry zone (DREZ), the point at which sensory axons enter the spinal cord. First, dorsal root injury does not compromise the blood–brain barrier, and spinal cyst cavity formation does not occur; second, astrocytes of the glia limitans form a precisely demarcated border between peripheral and central nervous tissue; and third, astrocytic and degenerative changes (microglial activation and myelin breakdown) are spatially and temporally separated from one another. Astrogliosis begins within days of a dorsal rhizotomy, whereas frank Wallerian degeneration does not begin until 1 week later.73, 74 It is clear that activated astrocytes, whether part of a scar following SCI or as a component of the reactive DREZ following rhizotomy, deter regeneration. Following SCI, axons retract from the lesion site and are surrounded by CNS myelin. They approach, but do not come into contact with the newly formed glia limitans at the innermost margin of the glial scar. This was demonstrated in studies by Silver and colleagues,75, 76 in which eGFP-expressing axons from grafted sensory neurons elongated within the degenerating dorsal columns, but halted within a halo of chondroitin sulfate proteoglycan (CSPG) immunoreactivity surrounding a small, previously generated, wound. Sensory axons injured within the dorsal root also regenerate up to the DREZ. Once there, their growth is arrested by the astrocytes of the glia limitans, against which they often form terminal-like axon structures,77 or from which they turn back, and are redirected peripherally.78
Based on observations such as these, it has been hypothesized that there exists an astrocytic ‘physiological stop signal’ at the DREZ which is responsible for the axonal arrest at the DREZ.79 While the nature of the signal(s) remains unknown, a number of ECM proteins, induced by dorsal rhizotomy, are well-situated to prohibit penetration by sensory axons of CNS tissue, including CSPGs, the CSPG NG2 made by oligodendrocyte precursors, as well as Tenascin-C and Tenascin-R.78 The astrocytic character of the inhibition strongly suggests secreted factors, and these are most likely to be components of the ECM laid down as a result of astrocyte activation. A recent DNA microarray carried out in our group to identify rapidly upregulated (3 days post-rhizotomy) astrocytic mRNAs has revealed a large number of candidate stop signals;80 here we review recent data on a selection of these molecules.
Chondroitin sulfate proteoglycans
A number of CSPGs are upregulated soon after dorsal rhizotomy, and are also candidate inhibitors of axonal regeneration at the site of SCI (reviewed in Morgenstern et al81 and Rhodes and Fawcett82). These include neurocan and versican, both of which are made by reactive astrocytes and inhibitory to neurite outgrowth.83, 84, 85, 86 While the growth-inhibitory effects of versican are well established,87 its pattern of expression following SCI is controversial,84, 86 and thus its role in the failure of regeneration remains unclear. Neurocan is more consistently upregulated after SCI, and thus this CSPG is a better candidate to mediate scar-related inhibition. While more slowly upregulated (only by 6 days post-rhizotomy), chondroitin synthase is likely to also play an important role in glial scar formation since it is involved in CSPG synthesis.88 The CSPG NG2 has been implicated in regenerative failure, based on its localization in the CNS part of the DREZ;78 in fact, its upregulation occurs too slowly (after 1 week) to compellingly implicate it in the rapid conversion of the DREZ from permissive to inhibitory (within 3 days) following rhizotomy.
Inhibitory effects of CSPG accumulation after SCI have been partially alleviated by enzymatic degradation. Chondroitinase ABC (ChABC), a bacterial enzyme that cleaves glycosaminoglycan (GAG) side chains from the protein core to attenuate CSPG inhibitory activity,89, 90, 91 promotes functional recovery when administered acutely following lesions of the cervical dorsal spinal cord in adult rats.92 Recently, ChABC was applied after thoracic hemisection, at the distal end of a Schwann cell-seeded polyacrylonitrile/polyvinylchloride (PAN/PVC) polymer channel, where it was reported to enhance axonal growth across the caudal graft–host interface.93 Although proven to increase plasticity in both spinal cord and brain,94, 95, 96, 97 ChABC does not completely remove GAG sidechains, and the ‘carbohydrate-stubbed’ protein core that persists after ChABC degradation of CSPGs does exert some inhibitory influence on growth of spinal axons.98 A DNA enzyme against the GAG-chain initiating enzyme, xylosyltransferase-1 (XT-1), may remove GAG chains more completely, and was recently applied to spinal stab lesions in adult rat:99 in this study, enzymatic treatment diminished GAG chain presence around the lesion and facilitated growth of GFP-positive axons from grafted sensory neurons beyond the lesion site.
Heparan sulfate proteoglycans (HSPGs)
Syndecan-1 is a member of the HSPG family identified in our microarray data. Four different syndecan genes have been identified, and all are upregulated with a rapid time course following brain injury.100 During development, the HSPGs are differentially expressed in lateral and medial astrocytes, with higher expression toward the midline thought to inhibit crossing of developing axonal projections.101, 102, 103 While the more medial astrocytes tend to inhibit axonal outgrowth, their treatment with heparatinase caused them to be more permissive to axonal elongation and rendered their surfaces (as measured with atomic force microscopy) more like the growth-promoting lateral astrocytes.104 A role for HSPGs in preventing inappropriate axon crossing at the optic chiasm, and in retaining retinal axons in the frog tectum has also been demonstrated.105
HSPGs also may act to increase inhibition by promoting astrocyte reactivity through an interaction with fibroblast growth factor (FGF) receptors:106 HSPGs increase the affinity of FGFs for their receptors. FGFs (particularly FGF-2) increase astrogliosis in vitro107 and in vivo,108 and they also increase the expression of syndecan-1 in oligodendrocytes and astrocytes.109 To date, few HSPGs have been identified at the DREZ, with the exception of glycipan, which is expressed in the E13-E16 DREZ,110 but not in the adult.111 The rapid upregulation of syndecan-1 revealed by the DNA microarray data is suggestive of a role for HSPGs in mediating inhibition at the DREZ and thus probably also in the damaged spinal cord, although HSPGs have not yet been targeted in animals models of SCI.
Keratan sulfate proteoglycans (KSPGs)
Lumican is one of the few cloned KSPGs.112 The induction of lumican in our DNA array data was surprising, given that its expression to date has been confined to cartilage and cornea.113 It is also noteworthy that lumican has been shown, using the Affymetrix DNA microarray, to be massively upregulated by pyramidotomy.114 This could mean either that lumican is de novo upregulated in reactive astrocytes or other CNS glia after injury, or equally plausibly, that there is sufficient sequence homology between lumican and other yet-to-be-cloned KSPGs to generate a false-positive result. In either case, the potential role of KSPGs in mediating axonal inhibition warrants discussion. Of the KSPGs so far identified in nervous tissue, claustrin,113 and ABAKAN115 are the best candidates for inhibiting axonal growth between CNS compartments. Claustrin was originally identified as a midline barrier molecule,116, 117 and the pattern of ABAKAN expression (separating thalamic nuclei, barrel fields and in astrocytes surrounding cortical stab lesions) is highly suggestive of a strong role in boundary formation.115, 118, 119 A recent report of KSPG induction following SCI has indicated that astrocytes are not robust producers of some KSPGs;120 however, the 5D4 antibody used has been reported to react with molecules other than claustrin and ABAKAN,121 and so these remain potential candidates for regeneration failure in the CNS.
ECM modifying proteins
A number of molecules involved in ECM synthesis and stabilization are also implicated by microarray data. Lysyl oxidase, while probably not astrocyte expressed, is rapidly upregulated following CNS trauma,122 and is known to catalyze the cross linking of ECM proteins.123, 124 Osteopontin (secreted phosphoprotein 1) is expressed by neurons, macrophages/microglia, and astrocytes following stroke125, 126 and ventral root avulsion injury.127 Its role in glial scar formation is suggested by its adhesive nature, as well as by its ability to promote astrocyte migration.128 Thrombomodulin is upregulated in activated astrocytes and may act to stabilize a glial scar following an initial astrocytic proliferative response induced by rhizotomy or direct CNS trauma.129 Two inhibitors of matrix metalloproteinases (MMPs) were also upregulated: tissue inhibitor of matrix metalloproteinase 1 (TIMP1) and alpha-2-macroglobulin (A2M). Both TIMP1 and A2M counteract the ECM-degrading properties of MMPs, leading to ECM reinforcement;130, 131 however, A2M has also been shown to have both astrocyte proliferation and neurite outgrowth-promoting properties.132
In contrast to the stabilizing effects of lysyl oxidase, osteopontin, thrombomodulin, and MMP inhibitors, a number of astrocytic proteins were upregulated which are known to have antiscarring properties. These include decorin, which when delivered to the site of a cortical stab wound, inhibits glial scar formation,133 and SC1, an antiadhesive glycoprotein synthesized and secreted by astrocytes.134, 135 It is likely that both stabilizing and de-stabilizing processes are operational in the first days following injury as the glial scar takes shape.
The cell body response and neurotrophic factors: upstaging intrinsic inhibition
Perhaps the most convincing studies demonstrating the requirement of an appropriate cell body response for axonal regeneration were carried out by Richardson and Issa,136 in which a prior lesion of the peripherally projecting axon of a dorsal root ganglion (DRG) neuron enhanced penetration into the CNS of a grafted centrally projecting branch. These experiments have since been repeated.137, 138 Of the various effects of such ‘conditioning’ lesions, two of the most important appear to be the upregulation of regeneration-associated genes (RAGs) such as GAP-43 and CAP-23 (reviewed in Bulsara et al139), and the upregulation of cAMP (reviewed in Qiu et al140). While the expression of GAP-43 does not necessarily imply that a neuron is regenerating,141 it does correlate well with the regenerative potential of a neuron or neuronal population. Even though GAP-43 is not upregulated following a dorsal rhizotomy or dorsal column crush lesion, a prior peripheral nerve injury both increases GAP-43142 and improves the sprouting and/or regenerative response to a subsequent central lesion.137
Neurotrophic factors are also well-recognized for their ability to induce RAG expression and to promote the elongation of axons. Following a dorsal rhizotomy, neurotrophin-3 (NT-3) application promotes functional centripetal regeneration as well as GAP-43 upregulation.74, 143, 144 Following dorsolateral funiculus lesions of the spinal cord, brain-derived neurotrophic factor application at the level of the red nucleus increases GAP-43 expression and improves regeneration of rubrospinal axons into a peripheral nerve graft.27
Like RAG expression, cAMP also appears to play an important role in regenerative potential. Cai et al40 demonstrated that if sensory or cerebellar neurons were pretreated with neurotrophic factors, there was a resulting increase in intracellular cAMP, which allowed axons to elongate on inhibitory substrates. Since then, the same group has shown that by augmenting intracellular cAMP in DRG neurons in vivo, central sprouting/regeneration is enhanced following dorsal column lesions.145 Intriguingly, the downstream effectors appear to be polyamines synthesized in part by Arginase 1.140 Both Arginase 1 and cAMP levels are high in young (neonatal) DRG neurons, correlating well with their ability to elongate in the presence of MAG.146, 147 Rolipram, a compound previously tested in clinical trials for treatment of depression,148 is a phosphodiesterase inhibitor, and thus causes cAMP to accumulate by preventing its enzymatic degradation. Rolipram has recently been administered systemically after SCI in rats, with encouraging results;149, 150 the central role of Rolipram in recent reports of combination approaches stimulating regeneration and functional recovery are discussed below (see ‘Casting the combination’).
Most studies examining the growth-enhancing effects of neurotrophic factors applied to the site of SCI have tested neurotrophins administered in conjunction with other manipulations. For example, nerve growth factor (NGF) can enhance axonal growth through fetal spinal cord transplants or peripheral nerve grafts.151, 152, 153 Additionally, BDNF and NT-3 enhanced growth of supraspinal and propriospinal axons into Schwann cell-seeded guidance channels in vivo,154 and boosted growth of descending axons into fetal transplants.155 Recently, NT-3 was reported to promote regeneration of dorsal column sensory axons into and beyond a graft of bone marrow stromal cells, but only when these neurons were preconditioned by cAMP injection into the DRG.156 Fewer studies have shown that treatment with neurotrophic factors on their own can induce axonal regeneration within the damaged spinal cord; however, intrathecally delivered NT-3 can induce regeneration of injured dorsal column axons.157 Neurotrophins have also been delivered to the site of SCI via genetically modified cells, which could conceivably play dual roles as both suppliers of trophic molecules and substrates for axonal growth. Fibroblasts, SCs, neural multipotent cells and olfactory ensheathing cells (OECs) have been modified to express various neurotrophins by ex vivo gene transfer; results of these important experiments have been reviewed recently.158
Since DRG neurons have been the best-characterized population in terms of responsiveness to neurotrophic factors, we will use them as a primary example. Significant inroads have been made into promoting re-entry of sensory axons into the adult spinal cord following dorsal root injury with these molecules (reviewed in Ramer et al159). Intrathecal NGF promoted the ingrowth of small-calibre axons that express CGRP, but not large-calibre axons that express NF200. By contrast, NT-3 promoted ingrowth of NF200-expressing axons, but not axons that express the P2X3 receptor (ie glial cell line-derived neurotrophic factor (GDNF)-sensitive axons).160 GDNF promoted the regeneration of large- and small-caliber axons, in line with the expression patterns of the requisite receptors.161, 162 Encouragingly, regrowth was accompanied by synaptic reconnection, again in a selective fashion, such that NGF and GDNF treatments resulted in reactivation of dorsal horn neurons by slowly conducting (smaller diameter) sensory axons, and NT-3 and GDNF treatments facilitated the recovery of postsynaptic potentials that are evoked by rapidly conducting (larger diameter) sensory fibers. NGF- and GDNF-treated animals regained the ability to sense pain, indicating that appropriate spinal circuits were being activated by sensory input. NT-3 promoted the appropriate recovery of proprioception,144 in-line with the distribution of trkC on proprioceptive sensory neurons. While much remains unanswered regarding the responsible mechanisms, effects other than those directly on neurons (such as nonspecific effects on glial cells) can largely be excluded as fundamental due to the specificity of the regenerative response and functional target reconnection.
Despite the robust regeneration obtained using neurotrophic factors in animal models, the use of neurotrophic molecules as potential therapeutic agents is not without some pitfalls. The most obvious example here is NGF, which while promoting the robust regeneration of small-diameter peptidergic afferents into the spinal cord following rhizotomy also produces severe hyperalgesia.163 One of the apparent issues is that NGF-treated axons far overshoot their targets164, 165 to terminate deep in the dorsal horn. Tang et al166 have taken advantage of a developmental mechanism of axon repulsion in the spinal grey matter by transfecting ventral cells with semaphorin 3A. This acts to prevent nociceptive axons from penetrating too deeply into the cord, and to partially inhibit the development of abnormal pain.166 Direct spinal cord trauma leads to the NGF-mediated sprouting of CGRP-positive fibers, which may be partly responsible for the development of autonomic dysreflexia (AD), a severe and life-threatening condition characterized by large and inappropriate elevations in blood pressure:167, 168 in this situation, sequestering NGF improves outcomes in rats and mice. By refining our ability to manipulate guidance cues, it may eventually be possible to extract from NGF its desired effects. For now, however, it remains an impractical member of the neurotrophin family for clinical administration after SCI.
p75: a wolf in sheep's clothing or a sheep in wolf's clothing?
Despite the well-characterized interactions of trks with p75 in neurotrophin signaling169 it remains unclear how p75 contributes to (or subtracts from) neurite outgrowth. In vitro studies using ciliary neurons have indicated that p75 activation by neurotrophins can increase trk-mediated neurite outgrowth: it has been suggested that by constitutively activating RhoA (a small GTPase involved in the control of actin filament assembly and focal adhesions), p75 impedes neurite elongation in the absence of neurotrophin. With the addition of ligand (such as NGF), RhoA activation is inhibited, promoting growth cone motility and neurite outgrowth.58 On the other hand, several studies have pointed to a negative role of p75 in neurotrophin signaling and neurite outgrowth: activating p75 in PC-12 cells with BDNF inhibited trkA activation through the ceramide pathway;170 in sensory neuronal compartmented cultures, binding of BDNF or a p75-specific antibody inhibited NGF-mediated trkA phosphorylation and slowed neurite outgrowth;171 and in vitro and in vivo, BDNF-mediated p75 activation inhibited sympathetic neurite outgrowth and target innervation.172 Additionally, in p75−/− mice in vivo, some brain structures are normally hyperinnervated,173 and regeneration following peripheral nerve injury is more robust in p75 knockouts than in wild-type mice.174, 175 In vivo, p75−/− sympathetic axons invade injured DRGs (where glial expression of NGF and NT-3 are increased176) more profusely than wild-type axons following a spinal nerve lesion.177 Other startling in vivo data argue strongly for a negative contribution of p75 to axonal growth in the CNS: in mice which overexpress NGF in CNS astrocytes, there is a massive developmental invasion of CNS white matter tracts by sympathetic axons, and this invasion was greatly augmented when the NGF overexpressors were crossed with p75 knockout mice.178
More recently, it has been suggested that p75 mediates inhibitory signaling of myelin-derived inhibitory proteins (see above).58, 59, 60 Upon binding to the common receptor for these proteins (the Nogo receptor), p75 is activated, leading to RhoA activation and inhibition of actin polymerization at the growth cone. On balance, these findings indicate a clear rationale for blocking p75 function in order to extend neurotrophin-mediated regeneration into or within the CNS following injury. Indeed, our group has recently found that central regeneration following dorsal rhizotomy is enhanced in p75−/− mice, as is intraspinal sprouting of descending monoaminergic (serotonergic, noradrenergic, dopaminergic) fibers.179 A recent publication showed that p75 antagonism did not promote the regeneration of injured dorsal column ascending (sensory) axons, but it noticeably prevented axonal retraction from the lesion site.22 It is interesting to note that ascending dorsal column axons also appear to be refractory to anti-NogoA treatment,180 indicating that the selection of a neuronal population for the study of regeneration is of critical importance in the development of potential therapies. It also argues for the investigation of multiple CNS pathways and at least the confirmation of any preclinical finding in another population, especially if the intervention is conceived as having a potential global therapeutic benefit for stimulating functional regeneration in patients.
A role for spared axons in functional recovery
Most SCIs are incomplete, and repair strategies should include the optimization of spared systems. In the absence of regenerative therapy, rodents exhibit functional recovery of complex motor tasks after SCI, and detailed analysis is often required to reveal deficits.181, 182 Axonal sprouting was inferred by Liu and Chambers,183 and has since been demonstrated repeatedly.114, 184, 185, 186, 187, 188, 189 This response of undamaged axons has been credited not only with producing motor improvements (reviewed in Muir and Steeves190 Dunlop and Steeves,191 and ‘Rehearsing roles’, below), but also with the instigation of chronic pain and AD.192
The ability of intact CNS axons to sprout remains largely unknown, but appears to depend on phenotype and the degree of cord trauma. NGF-sensitive axons sprout more than GDNF-sensitive axons following partial cord deafferentation.193 Intact descending systems (serotonergic and noradrenergic) respond differently to dorsal rhizotomy.194 Intrinsic dorsal spinal cord neurons will sprout into dorsal roots, but only when there has been (at least) some mild direct trauma to the spinal cord, as occurs following dorsal root implantation or insertion of a fine needle into the cord, but not after dorsal root injury alone.195, 196 That mild trauma to the cord promotes sprouting of intact axons suggests the involvement of neurotrophic factors and cytokines produced as a result of inflammation. There have been a limited number of demonstrations of neurotrophic factor-promoted sprouting of intact spinal axons. While NGF has been shown to promote intraspinal sprouting of nociceptive primary afferents,164, 165 the usefulness of NGF in SCI is severely limited by its painful side effects (discussed above).163 Exogenous NT-3 promotes sprouting of intact CST axons developmentally197 and following rubrospinal tract ablation in adults.184 Like neurotrophic factors, neutralizing inhibitory myelin and ECM molecules can promote sprouting of some intact systems: blocking Nogo-A promotes sprouting of adult CST axons,198 and digestion of glial scar-associated CSPGs promotes behavioral recovery following dorsal column injury in the absence of robust regeneration of injured axons.92
Bridging the site of SCI
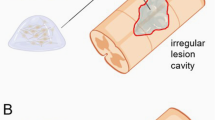
Neuroprotective treatments might soon increase the continuity across the lesion site (see ‘Neuroprotection in the setting of SCI’ above), but this advancement may not eliminate the need for bridging transplants. In addition to providing a permissive substrate for axonal elongation, the ideal bridge may confer continued growth support, remodel the lesion site to allow axons to pass through, lend protection from inhibitory molecules, and/or even stimulate remyelination. Here, we review recent data on axonal growth and behavioural recovery stimulated by cellular bridges tested in rats and humans. The use of cellular transplants to deliver neurotrophic factors (see ‘Stimulating axonal growth in the injured spinal cord’), to remove inhibitory debris at the lesion site (see ‘Stimulating axonal growth in the injured spinal cord’), or to stimulate remyelination (see ‘Overcoming conduction block’) is discussed elsewhere.
Classical experiments established that injured spinal axons can grow into a graft of peripheral nerve after complete transection of the spinal cord:32, 199 these seminal works initiated the search for the most suitable biological bridging agent. Peripheral nerves are still used experimentally to examine axonal growth in response to other manipulations, and can support some return of function after complete thoracic transection (in rat) with concomitant administration of growth factors.200, 201, 202 Peripheral nerves have been implanted at the site of SCI in patients in Taiwan, China, Peru, and Brazil and a report of such surgery recently became available.203 In this case, a patient with chronic paraplegia resulting from thoracic SCI was treated 4 years following injury with autologous sural nerve grafts and acidic FGF. In the 2 years following surgery, the patient exhibited motor and sensory improvements, indicated by the American Spinal Injury Association (ASIA) rating score (which changed from ASIA C to ASIA D) and pin-prick tests. However, data from a small study involving eight patients with functionally complete SCI who received similar treatment, presented at the International Clinical Trials Workshop on SCI earlier this year, was less encouraging.204 Autografts of peripheral nerve have also been used to bypass the lesion site: sural nerves grafted between ventral spinal cord above a thoracic lesion and ventral roots below restored some voluntary movement in one person with chronic SCI.205 Today, nerve bridges have largely given way to the development of cellular transplants for a number of reasons: single cells can be better-defined than the assortment of cells present in a whole nerve, and can be characterized in vitro; cells can be injected as a suspension, to fill all aspects of a cyst cavity at a lesion site, whereas implantation of a nerve segment may require resection; and finally, cells can be genetically modified to produce and release factors at the lesion site (see ‘Stimulating axonal growth in the injured spinal cord’ above).
In the spotlight: Schwann cells and OECs
Schwann cells (SCs) were the first to be cast as bridging agents for SCI. The rationale for injecting SCs into the injured spinal cord is clear, as SCs play a critical role in establishing the permissive nature for axonal regrowth within the peripheral nerve environment: when mitosis of host SCs is inhibited after sciatic nerve injury, axonal growth into acellular autographs is slowed.206, 207 After peripheral nerve injury, SCs act as ushers for growing axons by proliferating, decreasing their expression of various myelin proteins, and increasing the expression of neurotrophic factors and cell adhesion molecules.208, 209 Finally, Schwann cells can be obtained from adult peripheral nerve and rapidly expanded for autologous transplantation: human SCs have been successfully isolated, examined in vitro, and implanted into the injured rat spinal cord.210, 211, 212
Schwann cell transplantation at the site of SCI in the adult rat enjoys a prolific history and has been reviewed previously.213 Many experiments have tested a SC-filled PAN/PVC polymer channel, grafted at the site of acute thoracic transection in the adult rat.154, 211, 212, 214, 215, 216, 217, 218, 219 Without genetic modification or addition of neurotrophic factors, SC-filled grafts fuse with the cut stumps of the spinal cord, support ingrowth of propriospinal, sensory, and brainstem-spinal axons, and may underlie modest recovery of function, such as rhythmic stepping.213 However, supraspinal axons that enter SC bridges typically do not exit caudally to re-enter the spinal cord. The inability of descending axons to re-enter host tissue after complete transection may result from myelin inihibition and/or an accumulation of CSPGs at the distal graft-host interface,220 an obstacle which might be overcome with concomitant application of anti-inhibitory compounds and/or neurotrophic factors93 (see ‘Stimulating axonal growth in the injured spinal cord’ above).
In the early 1990s, another candidate for cellular bridging took the stage. OECs are glia that support growth of olfactory neurons – a routinely replenished population of neurons – from PNS-to-CNS throughout adult life. The rationale for implanting OECs into the injured spinal cord is less obvious, since when the axons of olfactory neurons are injured, they do not regrow their axons like peripheral nerves, but rather rapidly degenerate and die.221, 222, 223 New neurons reconstitute the injured peripheral olfactory system,224 but do not restore original connectivity, and this is reinforced clinically: permanent anosmia or dysosmia are relatively common after blunt head injury, when movement of the brain relative to the skull shears olfactory axons at the cribriform plate.225 The rationale for using OECs, then, is not that they support axonal regeneration in situ, but that they exist in a unique capacity to permit axonal growth across a PNS–CNS interface in the adult.
Since olfactory axons grow across a PNS–CNS interface, OECs (from the rat olfactory bulb) were first injected into the rat spinal cord after dorsal root injury, to examine their ability to bridge the DREZ.226 In this study, afferent ingrowth was reported to extend into the dorsal horn, in the areas of appropriate targets, and subsequent electrophysiological experiments suggested that these axons achieved functional reconnection.227, 228, 229 However, recent data from our group and others indicate that OEC transplantation does not support regeneration of sensory axons across the injured DREZ.230, 231, 232 Using OECs from the GFP mouse, we have observed that OEC injection into the spinal cord mechanically disrupts the DREZ, permitting sensory axon ingrowth but not functional regeneration involving the re-acquisition of appropriate targets on the other side of the injured DREZ.232
After SCI, OECs were also applied to remodel a PNS–CNS interface, namely, that established by grafting Schwann cells into CNS tissue. When OECs were injected into the proximal and distal stumps of transected spinal cord spanning a SC bridge, regenerating axons crossed the graft–host interfaces and re-entered the spinal cord.233 Other experiments indicated that OECs alone were sufficient to support both axonal growth and functional recovery.234, 235, 236, 237, 238 These data have generated sufficient enthusiasm in the SCI community to instigate several clinical trials testing OECs worldwide. The first report of a large trial in China was recently published.239 Although OEC transplantation was reported to generally improve both sensory and motor function, clinical follow-up was limited, confounding interpretation of these results. As well, OECs in this trial were harvested and expanded from the fetal olfactory bulb, an ethically problematic cell source. In Lisbon, Carlos Lima has grafted pieces of autologous olfactory mucosa at the site of SCI in 20 patients, without intervening in vitro manipulation or isolation of OECs; some return of both sensory and motor function has been reported after these surgeries.204 Recently, OECs have been successfully isolated and expanded from the adult human olfactory mucosa, accessible by nasal biopsy, potentially permitting autologous transplantation:240 these cells have entered a small Phase I trial in Brisbane, Australia.
As OECs are being clinically tested, work in animal models continues, both to identify potential mechanisms of OEC-mediated recovery and to optimize methods of OEC delivery. Recent data suggest that the mechanism of functional return may be more complex than regrowth of lesioned axons. In fact, OEC transplantation protects spinal tissue from secondary damage and prevents cavitation,241, 242, 243, 244, 245, 246 enhances vascularization of the lesion site235, 246 and promotes branching of neighboring axons spared by the primary injury,247 all of which might subserve improved functional outcome. While earlier transplant experiments sought to isolate OECs without other cellular components of the olfactory nerve, recent work suggests that the recovery of function is enhanced by including other cell types, such as olfactory nerve fibroblasts (ONFs), in the graft.248 Grafts of OECs and ONFs were recently shown to reconstitute ipsilateral breathing rhythm in rats with cervical hemisections,249 although functional recovery after spinal hemisection must be interpreted with caution, as it has been well-documented that both hemisected people and animals experience substantial and spontaneous recovery via preserved (undamaged) projections. Finally, the timing of OEC transplantation may be important. Recent data indicates that OEC transplantation has functional benefits even when delayed (for 1 week to 2 months after injury in the rat)250, 251 and one study suggests that delayed transplantation is actually more beneficial than acute delivery.244 To address some of these important issues, and to more confidently predict the effects of OEC transplantation in humans, OEC transplantation is being investigated in non-human primates with complete spinal cord transection (A. Ramon-Cueto, pers. comm.).
SCs and OECs: a duel or a duet?
Despite the relatively rapid progression from animal models to clinical trials, OECs have not clearly outperformed SCs as cellular bridging agents. The two cell types are comparable in some respects, but not in others.252 Like SCs, OECs express adhesion molecules and neurotrophic factors and can support growth by many types of neurons.152 Unlike SCs, OECs do not proliferate or migrate in response to injury of their associated axons.253 Both types of cells have been genetically modified prior to transplantation to deliver growth factors at the lesion site.245, 254, 255 OECs have been reported to integrate more successfully with astrocytes,256 and to induce less glial scarring257 than SCs, but may themselves express inhibitory molecules (such as Nogo or proteoglycans, discussed in ‘Stimulating axonal growth in the injured spinal cord’ above) after transplantation.258 Few experiments have directly compared SC- and OEC-transplantation in the same injury. When SCs and OECs were injected (separately and together) into acute thoracic contusion injuries in rat, both cells induced tissue sparing and angiogenesis at the lesion; however, functional outcome was only improved in animals that received SCs.243 SCs were also recently reported to support regeneration of rat retinal ganglion axons, while OECs applied in the same manner did not.259 In a recent combination study, SCs bridged a thoracic contusion site to support growth and remyelination in rolipram and cAMP-treated animals (see ‘Casting the combination’, below).260
On a final note, OECs and SCs may share the stage: recent work from our group demonstrates that OEC transplantation enhances invasion of host SCs into the injured spinal cord.246 This finding is especially significant in light of recent data indicating that myelin within OEC grafts is SC-, not OEC-derived261 (discussed in the subsequent section below). Whether the observed axonal growth and/or functional recovery after SCI can be attributed to the presence of grafted OECs or might also be due to infiltrating host SCs remains to be seen. SC infiltration/penetration of the CNS has been linked to regeneration of spinal262, 263 and vagal afferents.264
In the wings: fetal tissue, multipotent/progenitor cells and synthetic implants
Spinal transection in the vertebrate embryo, prior to the onset of myelination, results in functional regeneration and repair.265, 266, 267, 268 Akin to the use of peripheral nerve grafts, grafts of fetal brain and spinal cord have been applied to the site of SCI to provide axons in the adult spinal cord with the permissive environment of the embryo269, 270, 271 (reviewed in Anderson et al272 and Lakatos and Franklin273). In addition to acting as a permissive substrate for elongating host axons, grafted embryonic neurons can both receive synaptic input from and extend axons to host spinal neurons, thereby acting as a relay across the injured spinal cord.274 Fetal tissue can be used to replace some of the neurons lost in SCI: for example, pieces of ventral embryonic spinal cord can give rise to neurons that reinnervate muscular targets and reverse muscle atrophy in rat.275 Analagous to the peripheral nerve environment, axons grow readily into grafts of fetal tissue, but emerge distally in a more limited fashion, a limitation that may be overcome by concomitant administration of neurotrophic factors.155, 276 Transplantation of fetal spinal tissue has been tested clinically in the United States in patients with progressive post-traumatic syringomyelia, and preliminary reports suggest that this treatment is safe,277, 278 although complete reports have not been published. Since the use of fetal tissue entails ethical challenges, it may need to be proven superior to other biological bridges in order to assume the role of a preferred clinical bridging intervention.
Recently, various populations of multipotent and progenitor cells have gained increasing attention as potential grafting agents for SCI. Spinal and supraspinal axons grow into grafts of neural multipotent cells, indicating that they do function as a permissive bridge,279, 280, 281 but the possibility that multipotent/progenitor cells might differentiate into spinal neurons to replace lost spinal circuitry is perhaps more intriguing. Both embryonic multipotent cells and neural progenitor cells can differentiate into neurons and extend axons into host tissue,26, 282 raising the possibility that multipotent cell grafts could also function as a relay center within the injured spinal cord. To date, however, neuronal differentiation of multipotent cells grafted at the site of SCI has been limited, and the vast majority of grafted toti- or mulitpotent progenitors become glia.283, 284 Interestingly, functional recovery after thoracic hemisection was improved when neural multipotent cells were delivered in a degradable polymer scaffold, despite the fact that grafted cells did not differentiate into neurons.285 In this study, improved functional outcome was associated with tissue sparing and reduced glial scarring, suggesting that multipotent cells may exert protective effects. Improved functional recovery from multipotent-cell-treated SCI associated with oligodendrocyte differentiation and remyelination of host axons is discussed as a potential remyelination strategy (in next section).
Multipotent cell-induced recovery in animal models has prompted clinical testing in Russia, where 15 patients received injections of fetal brain and hemapoietic (liver) cells; 11 patients received concomitant grafts of OECs and fetal spinal cord.286 In this study, six patients that exhibited complete sensory and motor paralysis prior to transplantation regained the ability to walk with or without assistance. However, five patients in this group received transplants within 4 months of SCI, and four received OECs and fetal spinal cord in addition to multipotent cells, so the results of this study are difficult to interpret. While no complications related to multipotent cell transplantation have been reported, the incomplete understanding of multipotent cell regulation, proliferation, and differentiation both in situ and upon transplantation may have important implications for imminent clinical translation of this technology.287, 288
Despite its promise in animal models, cellular transplantation presents several challenges for clinical translation. Timing of transplantation, cell source, and cellular maintenance/manipulation are important inherent variables that may preclude comparison of results from clinical trials at different centers. Additionally, immunological compounds may be associated with graft rejection. Synthetic implants under development offer the attractive potential of being identical and reproducible in composition, immunologically inert, and even absorbable. If cellular transplantation after SCI does become a clinical reality, synthetic implants may be used to contain/deliver the cells at the injury site and/or to guide axons growing through these biosynthetic grafts.213 Recently, biomaterials have been designed to deliver neurotrophic factors, and may provide an attractive alternative to the intrathecal pump, as delivery is reproducible and immobilized concentration gradients can be created.289, 290, 291 In two recent experiments, synthetic implants were applied to the site of complete thoracic transection in rat; in these studies, BDNF-filled foam292 and synthetic hydrogel guidance channels293 integrated with host tissue and supported axonal ingrowth, although axons did not regenerate beyond the implant. While we want to stress the potential importance of synthetic implants in stimulating regeneration and recovery after SCI, we are constrained by space, and direct the interested reader to reviews of developments in this field.294, 295, 296
Overcoming conduction block
Although the CNS has the capacity to replace oligodendrocytes lost in spinal trauma, and remyelination of intact demyelinated axons can be spontaneous,297, 298 remyelination in the wake of SCI is not robust, and the resulting conduction block of action potentials due to persistent demyelination likely contributes to loss of function in SCI.299 To supplement intrinsic CNS remyelination, myelinogenic and progenitor cells have been grafted at the site of SCI in rats, and such cells have been tested in both focal demyelinating lesions (inducing oligodendrocyte death without axonal trauma) and traumatic lesions (resulting in axon damage). Remyelination may differ among intact versus regenerating axons; nonetheless, models of focal deymelination have provided important information regarding the extent and mechanisms of spinal remyelination stimulated by cell transplants. Here, we review recent data in this field, and also consider another approach to alleviating conduction block via systemic drug treatment, which is currently being tested in a clinical trial.
Stage left: Schwann cells and OECs
In addition to the fame they have acquired in supporting axonal elongation (see ‘Bridging the site of SCI’), SCs and OECs have also been acclaimed for their ability to remyelinate axons after SCI. Endogenous SCs can invade spinal lesions and form functional myelin,300, 301 providing the impetus for transplantation of SCs (and by association, OECs) to alleviate SCI-induced conduction block. Experiments reporting SC- and OEC-provoked remyelination after spinal insult have been reviewed recently;302, 303, 304 therefore, we limit this discussion to recent data that implicates SCs as a source of peripherally derived myelin after both SC- and OEC-transplantation into spinal lesions.
OEC myelination was first reported in vitro. When rat OECs were cocultured with rat DRG neurons or olfactory receptor neurons, myelin sheaths formed that were indistinguishable from those formed by SCs under similar conditions.305, 306 In a recent study, however, OECs cultured from adult rats did not retain the ability to myelinate embryonic DRG neurons; instead, myelin in DRG cultures was formed by Schwann cells that persisted in culture despite antimitotic treatment with fluorodeoxyuridine (FUDR).307 These data, demonstrating the propensity of SCs to myelinate axons even in the presence of OECs, have potentially important implications for interpreting the results of OEC transplantation experiments.
Focal areas of demyelination in the adult spinal cord can be created by injections of ethidium bromide or lysolecithin, both of which are toxic to oligodendrocytes. Typically, the demyelinating lesion is created in tissue that has been X-irradiated several days previously, which prevents host oligodendrocytes from repopulating the lesion site.308 Using this model, peripheral myelin has been observed in the demyelinated dorsal columns of the adult rat subsequent to transplantation of rat, human, and canine OECs309, 310, 311, 312, 313, 314 and recently, in the dorsal columns of the monkey following porcine OEC transplantation.315 In these experiments, peripheral myelin was morphologically and/or antigenically indistinguishable from that formed by SCs myelinating previously demyelinated CNS axons.316, 317
When OECs and SCs were grafted at the same type of contusion injury, more myelinated axons were found within SC-containing grafts than within OEC-only grafts.243 Importantly, peripherally myelinated axons were also found in nontransplanted lesions, demonstrating that host SCs could access the untreated contusion site, in limited numbers, and produce myelin.243 Remyelination has also been achieved by stimulating Schwann cell immigration via cellular delivery of GDNF at the site of thoracic transection.318 Recently, Boyd et al261 used virally labeled OECs to demonstrate that peripheral myelin that formed within the OEC-transplanted site of SCI was derived entirely from invading host SCs and not from grafted OECs. Taken together, these provocative data highlight the role of endogenous SCs in accomplishing remyelination of spinal axons. While it has been reported that SC remyelination of spinal axons is a transient phenomenon, with SC myelin progressively replaced by oligodendrocyte myelin during recovery,319 the most recent data indicates that SC myelin is stable and persists in the spinal cord.
Stage right: progenitor cells
In response to demyelination of spinal axons, a resident population of endogenous oligodendrocyte precursors proliferate and differentiate into mature oligodendrocytes to initiate remyelination.320, 321, 322 Progenitor cells have been grafted at the site of SCI to enhance this repair process. While grafts of SCs or OECs trigger formation of peripheral myelin, progenitor cell transplantation aims to reconstitute central myelin at the site of SCI. Neural progenitors from both mouse embryonic multipotent cells and embryonic rat spinal cord have been injected at the site of contusion injury in rat, where both types of grafted cells differentiated into oligodendrocytes (as well as astrocytes and neurons) and enhanced functional recovery,323, 282 although remyelination was not examined in either study. Stem-like cells, derived from bone marrow, have been reported to myelinate spinal axons when introduced at the site of focal demyelination.324, 325 Intriguingly, these cells also stimulated remyelination – and themselves formed myelin – when injected into the bloodstream.325, 326 If such cells can reliably overcome the blood–brain barrier, migrate to demyelinated CNS regions and differentiate appropriately, with their proliferation being controlled, then an intravenous delivery route presents an attractive alternative to direct parenchymal injection into the damaged CNS.
Now showing: treatment with 4-aminopyridine
While cellular transplantation might rebuild stable myelin to permanently restore conductivity, transplantation procedures are invasive and immunologically problematic. Pharmacological treatment, although it must be chronically sustained, may represent a more imminent solution to the problem of conduction block. Fampridine, or 4-aminopyridine (4-AP), is a potassium channel blocker that restores conduction in de- or dysmyelinated axons.327, 328 When 4-AP is added to an in vitro preparation of injured spinal cord, conduction across the site of SCI is improved.329, 330, 331 In people with incomplete SCI a sustained-release (oral) formula of 4-AP (Fampridine) was reported to improve sensory and motor function and reduce spasticity during treatment.332 As a result of these encouraging data, Acorda Therapeutics (New York, USA) initiated two large, multicenter clinical trials testing 4-AP in chronic SCI (8) and published results of these trials may soon become available.204 Animal experimentation with 4-AP is ongoing, in an effort to clarify the optimal parameters of 4-AP treatment. Although pharmacological intervention is logistically simpler than cell transplantation, issues of effective dose and delivery may be complex; for example, recent data revealed that 4-AP more potently overcomes conduction deficit induced by stretching forces than conduction deficit induced by compression.333 These experiments underscore the importance of animal models in optimizing treatments for SCI, even and especially after clinical trials are underway.
Rehearsing roles: rehabilitation and CNS plasticity
Even though there is little evidence for spontaneous de novo regeneration of damaged CNS axons over substantial distances, there is overwhelming evidence for short- and long-term changes within existing circuits.334, 335 The extent to which such plasticity is accomplished as a result of axonal sprouting or short-distance regeneration versus compensatory changes within existing circuits remains to be determined.190, 336 Nevertheless, the available studies point to an innate and remarkable ability for the damaged adult CNS to undergo spontaneous or activity-dependent plastic changes. A choreographed recovery after SCI is dependent on a diverse number of players, contributing their parts in many subtle, but important, ways. Recent research has been able to identify a diverse number of poorly understood mechanisms that can contribute to a remodeling of the CNS after injury. The challenge for rehabilitation after SCI is to ensure that these alterations are beneficial rather than detrimental and maximized to their fullest extent.
For example, damage to the periphery (eg, by digit amputation, retinal lesions) results in latent responses being recorded almost immediately (within minutes) from cortical regions that had previously received input from the damaged regions.335 In other words, loss of input from the periphery can result in the rapid appearance of expanded receptive fields of the remaining cortical neurons. This phenomenon has been referred to as synapse ‘unmasking’, a process proposed by Basbaum and Wall to underlie receptive field expansion following partial spinal deafferentation.337 Later, an anatomical correlate of synapse unmasking was described by Goshgarian et al, which occurred within 4 h of SCI, and involved astrocytic process retraction and a subsequent increase in the area of synaptic apposition. The rapidity of the changed response patterns revealed a previously under appreciated feature of CNS organization, namely that each region receives input from a much wider peripheral area than can normally be detected electrophysiologically.334, 335 Furthermore, the data suggest that much of the input to the receptive field must normally be subthreshold for detection or actively suppressed (inhibited) and that damage brings about an almost immediate expansion of the receptive field.
In addition to short-term plastic activity changes, long-term changes also occur and appear to be underpinned by anatomical sprouting or rewiring of synaptic connections (see above338) Small binocular retinal lesions initially silence the corresponding portion of the visual cortex. However, within weeks to months, the cortex becomes responsive to inputs from adjacent portions of the uninjured retina. Anatomical tracing studies revealed extensive axonal sprouting from the adjacent cortex, which was interpreted as arising from intrinsic neurons that had formed new stable connections within the denervated region. Likewise, recent reports using trans-synaptic tracing techniques, electrophysiology and behavioural assessments have demonstrated a significant degree of plasticity involving compensatory sprouting from corticospinal projections with concomitant functional rewiring after a dorsal hemisection injury of the adult rat thoracic cord.339 It is important to recognize that even if regeneration of injured axons is successful, or if synaptic spaces can become newly occupied by intact axons, the target in both cases is profoundly altered by injury. As a consequence, functional reovery will inevitably require some form of retraining.
Practice makes perfect
Given the innate capacity for the damaged nervous system to undergo plastic changes, the question arises as to whether plasticity within undamaged CNS circuits or regenerating pathways can be harnessed to improve functional outcome. Reports point to the importance of appropriate training to enhance recovery after damage. After sciatic nerve crush in rats, both sensory and motor recovery is improved by placing water bottles at heights such that animals have to extend both hind paws maximally to drink; animals with water bottles on the floor do not show improved recovery.340 There are, however, offsetting data in the same report suggesting that overactivity or stressful regimes such as forced running or swimming can impede rather than enhance functional recovery.
Generation of movement requires the coordination of a vast network of neuronal subsets, resulting in the coordinated activation of the limbs. There is ample evidence that the intrinsic (or isolated) spinal cord circuitry has the capacity to directly generate many of the complex aspects of locomotion. Complex motor patterns are not just programmed by supraspinal systems such as the cortex, basal ganglia, and cerebellum, with the spinal cord contributing only passively or reflexively by relaying sensory and motor information to the brain or muscles.341
Locomotion, whether walking, running, flying, or swimming, is ultimately produced by spinal neurons that are collectively termed central pattern generators (CPGs). Normally, CPGs are activated by supraspinal control from the brainstem and thalamus and are also continuously modified by peripheral cutaneous and proprioceptive input. However, it has long been known that the spinal cord can act independently to produce locomotor movements. Indeed, CPGs appear to be a broad vertebrate characteristic because removal of supraspinal input in lampreys, birds, nonprimates, and primates (including humans) by complete or nearly complete spinal cord transection results in the retention of locomotor movements.342, 343, 344 In these instances, locomotor movements can be initiated by a variety of stimuli such as certain postures, electric shocks, or exercise. Given that the spinal cord has the capacity to encode and execute both stepping movements and higher-order functions associated with locomotion, the evidence is now accumulating that these abilities can be harnessed to generate beneficial function after spinal injury.
Training and neural activity to harness spinal cord plasticity
The cat has proved to be an extremely valuable model for assessing the effect of training on the ability of the isolated spinal cord to generate stepping movements after complete spinal cord transections, which were undertaken at low levels, namely T12–13. It has now been firmly established that, with interactive training, adult spinal transected cats can rapidly develop locomotion. Initial studies showed that animals developed stepping patterns on a treadmill if their weight was partially supported and their balance stabilized.345 Strong pinching of the perianal and abdominal regions was only necessary in the first few days of step training and could be reduced to very light stimulation over the following 3 weeks. All aspects of locomotor behavior improved over the subsequent months, and animals were able to support their body weight, often stepping on the treadmill for up to 10 min before losing their balance. However, without daily step training, performance regressed with animals dragging their hindlimbs, although locomotor performance could be re-established within a few days by step training.
The improved performance compared to spontaneous recovery was attributed to neurons being activated in a more appropriate fashion by training. Recent data obtained using a rodent exercise device indicated that a daily exercise program of moving paralyzed hindlimbs through the motions of walking prevented atrophy of spinal motor neurons.346 Furthermore, it was suggested that neural activity facilitated the return of function in existing sensorimotor pathways rather than the generation of new pathways. Although some spontaneous recovery was seen when animals were not step trained but were tested briefly on the treadmill, these animals showed only short bouts of weight-bearing stepping and stumbled frequently.347
If the isolated spinal cord can learn specific tasks, how long can it remember them? Cats that underwent treadmill step training for 12 weeks still performed maximally on all criteria when tested on the treadmill for up to 6 weeks after cessation of training.348 However, performance declined considerably if training was withheld for 12 weeks. Nevertheless, retraining after 12 weeks of withdrawal resulted in a more rapid improvement in stepping than that seen during the initial training period. The results suggest that the isolated spinal cord can learn, and it will forget a task without maintained use or practice; however, the isolated spinal cord retains trace memories that can be rapidly recovered upon renewed training.
For the majority of individuals with functionally complete injuries, there is a small band of spared tissue that bridges the gap. Thus, clinically (functionally) complete lesions are not necessarily ones where anatomically the spinal cord has been completely severed. Studies have shown that some locomotor recovery can occur in animals where only a small subset of axons, spanning a lacerating lesion, is spared.349, 350 In addition, after an initial contusion injury that spares some axons around a central cavity and that mimics the type of closed SCI most commonly seen in humans, rats recover some locomotion.351 The surprise was that if the spinal cord was then completely cut, rats were still able to maintain some locomotor function, something that rarely happened after spinal cord transection alone. Furthermore, animals recovered a greater range of motor movements and did so more rapidly than after transection alone. The interpretation was that even small percentages of spared axons are capable of ‘instructing’ the spinal cord below the lesion and that the instructions are retained.
Treadmill training has been used with considerable success in people with SCI, classified as functionally incomplete,352 that is, with retention of some sensory or motor function below the level of injury (ASIA scale B–D). Persons were selected if they had some voluntary activity in leg muscles, had mobile joints, had no spasticity and if they lacked complications such as ulceration or infection. Treadmill training involved sessions of 30–60 min, 5 days a week for 3 weeks to 5 months, starting with low treadmill speeds. The aim was to encourage movements that mimic natural walking as much as possible and involved providing maximal sensory feedback from the muscles, joints, and skin.
Initially, partial body-weight support was provided by a harness, and the legs were moved and the feet were placed on the treadmill by physiotherapists. Persons were encouraged to put their full body weight onto the extended (say, right) leg during the stand phase and then shift their body weight onto the left leg just before swinging the right leg forward. As performance improved, persons were encouraged to swing their arms in the way they would if walking naturally. Persons were also encouraged to attempt overground walking as soon as possible. The study included comparison with persons who had previously received conventional physiotherapy only. Treadmill training, rather than conventional physiotherapy, resulted in remarkable improvements. Of 44 patients that were wheelchair-bound, over two-thirds learned to walk independently, at least for short distances, with only one person not showing any improvement; about half of these patients were capable of walking up stairs. Seven paraplegic persons classified as complete, that is, no function below the level of injury, were also included in the study and were selected because some aspects of stepping could be evoked by placing them on the treadmill. However, despite daily training, no improvements were seen, and they were not able to generate full stepping cycles. Follow-up studies in persons with incomplete injuries were encouraging and showed that improvements made during a few weeks to months of treadmill training were maintained for 6 months to 6 1/2 years after training ceased.352, 353
The long-term effects of treadmill training have recently been compared for individuals that are functionally incomplete and complete.354 After training (15 min daily, 5 days per week for several months), persons with incomplete injuries showed significant improvements and could use their new locomotor capabilities for overground walking for at least short distances. The benefits that had been acquired during training could thus be maintained by independent movement, although there did not appear to be any further improvements. However, all individuals with complete injuries were unable to maintain stepping movements after training stopped; thus, the stepping that had been learned during training could not be translated into overground walking and furthermore appeared to be lost in the absence of continued practice (‘use it or lose it’).
Rehabilitation rigor
Rehabilitation research often lacks a foundation of experimentally derived data. There have been few multicenter trials including a statistically significant number of participants with double-blind assessments and randomized controls. Rehabilitation has been susceptible to the practice of procedures developed on the basis of personal experiences of the therapist, in part because there are few catastrophic consequences if a therapeutic strategy fails. All too often, it has also been difficult to objectively measure a beneficial outcome or standardize an effective treatment strategy across multiple rehabilitation centers that is independent of the important, but confounding trial variable, ‘chemistry’ that intrinsically develops within a therapist–client relationship.
However, this is changing rapidly. For example, there is currently a multicenter NIH-sponsored trial, using blinded assessments of people with incomplete SCIs, investigating the benefits of weight-supported treadmill training versus more conventional forms of active physical therapy. Other active forms of rehabilitation such as constraint-use therapy and functional electrical stimulation are also undergoing more rigorous evaluations.
In summary, neural activity shapes the developing and adult CNS. If people with SCI can regain some function through the myriad plasticity changes in whatever small number of undamaged pathways persist after injury, then the possibility of combining active rehabilitation therapy with surgical and therapeutic interventions promises realistic opportunities for improved functional recovery after SCI in the near future.
Casting the combination
Several recent studies highlight both the potential success of combination therapy, and the complexity inherent in its development. These studies combine growth-promoting and bridging strategies in three different models of SCI in the adult rat. They are notable not only because the results are encouraging, but because they represent collaborative efforts between experts in different fields, the type of cooperation required to develop a successful combination therapy for SCI.
In one study, sensory neurons were primed for growth in some animals by injecting dibutyryl-cAMP (db-cAMP) into the lumbar DRGs 5 days prior to a cervical lesion of the dorsal columns.156 Immediately following the injury, all animals received implants of autologous bone marrow stromal cells (BMSCs), and some animals received injections of NT-3 at the lesion site. At 1 week following injury, some animals received injections of NT-3 rostral to the lesion. In order to dissect the effects of each treatment, this study included five experimental groups. While sensory axon growth into the BMSC graft was improved by any combination of cAMP and/or NT-3 injections, significant growth beyond the graft was only observed in animals that received both cAMP injections prior to surgery and NT-3 injections at and beyond the lesion following injury. No significant recovery of function was observed in any of the animals, likely because regenerating sensory axons did not reach their targets in the nucleus gracilus. Although stimulating regeneration through preconditioning is not clinically applicable, this study illustrates that growth-promoting interventions aimed at different parts of the neuron – the axon and soma – can have additive effects.
In another study exploiting the growth-promoting effects of cAMP, a cervical hemisection was treated with embryonic spinal tissue, and (in experimental animals) rolipram.149 The graft was implanted immediately following injury, and rolipram was administered 2 weeks later, for a duration of 10 days. Rolipram-treated animals exhibited both increased axonal growth into the embryonic transplant and evidence of improved forepaw placement compared to vehicle-treated controls. While this experiment tested the efficacy of different concentrations of rolipram, it did not examine the effects of rolipram treatment alone, without concomitant grafting, or of administering rolipram immediately following SCI.
The effectiveness of acute rolipram was examined in a third study, in which a moderate thoracic contusion injury was treated with various combinations of a SC graft, a single injection of db-cAMP near the graft, and systemic administration of rolipram over 2 weeks.150 The timing of intervention is of interest in this experiment, as rolipram treatment was initiated either at the time of injury or 1 week following injury, while SC grafts and db-cAMP injection were administered only 1 week following injury. Even without testing all combinations of acute versus delayed interventions, this study involved seven different experimental groups. Rats that received rolipram immediately following injury plus SC grafts and db-cAMP exhibited increased sparing and growth of supraspinal axons, as well as improved locomotor recovery, compared to rats that received only SC transplants; delaying rolipram administration generally reduced treatment efficacy. By most outcome measures reported, the combination of SC graft plus acute rolipram also enhanced axonal sparing/growth and functional recovery. Interestingly, acute rolipram alone was sufficient to augment both sparing/growth and functional recovery relative to untreated controls.
While combination therapy has achieved star status in recent years, attempts to synergistically combine treatments aimed at different components of SCI have been ongoing for more than a decade. Table 1 summarizes many (but by no means all) of the attempts at combination therapy applied in adult spinal cord. In addition to the sheer number of studies combining different treatments, the distribution of combinations are of interest. For example, cell transplants and neurotrophic factors have been combined many times, while combinations testing rehabilitative treatments are comparatively scant. This is unexpected, given that most SCI patients have access to some form of rehabilitation and that any treatment that moves to clinical trial is likely to be tested on a background of some form of rehabiliation. A review of combination therapies also serves as a warning that unforseen interactions between treatments may preclude the application of some in combination. For example, a recent study revealed that acute delivery of methylprednisolone and interleukin-10 worsened the behavioral outcome of SCI in animals that received SC transplants.260
The complexity of combination therapy will only escalate in prospective clinical trials where randomized control groups and blind assessment will be required to test safety and efficacy of candidate therapies. Some components of SCI therapy may entail less risk, and thus move to clinical trial sooner, than others. For example, pharmacological treatments may entail less risk than surgical interventions, although both may be required to achieve recovery after SCI. Such risk–benefit considerations will vary between patients according to the type, level, and extent of injury to further complicate approval for and analysis of clinical trials. These and other issues surrounding clinical trial design, including an urgent need for communication between regulatory agencies and the SCI research community, were raised at the International Clinical Trials Workshop on SCI held in Vancouver in 2004.204
Despite these intrinsic difficulties, the show must go on, and clinical trials must be guided by animal experiments that rigorously test neuroprotective, growth-promoting, bridging, remyelinating and rehabilitative therapies for SCI in isolation and in combination. The successful combination therapy will consider the interactions between players to ensure that they take the stage at the correct place and time to maximize efficacy and minimize risk, and will reveal that there are, truly, no small parts.
References
Bunge MB . Bridging areas of injury in the spinal cord. Neuroscientist 2001; 7: 325–339.
Hausmann ON . Post-traumatic inflammation following spinal cord injury. Spinal Cord 2003; 41: 369–378.
Bracken MB et al. Methylprednisolone or tirilazad mesylate administration after acute spinal cord injury: 1-year follow up. Results of the third National Acute Spinal Cord Injury randomized controlled trial. J Neurosurg 1998; 89: 699–706.
Geisler FH, Coleman WP, Grieco G, Poonian D . The Sygen multicenter acute spinal cord injury study. Spine 2001; 26: S87–S98.
Bracken MB et al. Methylprednisolone or naloxone treatment after acute spinal cord injury: 1-year follow-up data. Results of the second National Acute Spinal Cord Injury Study. J Neurosurg 1992; 76: 23–31.
Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR . Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J 2004; 4: 451–464.
Hugenholtz H . Methylprednisolone for acute spinal cord injury: not a standard of care. CMAJ 2003; 168: 1145–1146.
Blight AR . Miracles and molecules – progress in spinal cord repair. Nat Neurosci 2002; 5(Suppl): 1051–1054.
Bethea JR, Dietrich WD . Targeting the host inflammatory response in traumatic spinal cord injury. Curr Opin Neurol 2002; 15: 355–360.
Schwartz M, Lazarov-Spiegler O, Rapalino O, Agranov I, Velan G, Hadani M . Potential repair of rat spinal cord injuries using stimulated homologous macrophages. Neurosurgery 1999; 44: 1041–1045.
Blight AR . Delayed demyelination and macrophage invasion: a candidate for secondary cell damage in spinal cord injury. Cent Nerv Syst Trauma 1985; 2: 299–315.
Shuman SL, Bresnahan JC, Beattie MS . Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res 1997; 50: 798–808.
Popovich PG . Immunological regulation of neuronal degeneration and regeneration in the injured spinal cord. Prog Brain Res 2000; 128: 43–58.
Jones TB et al. Passive or active immunization with myelin basic protein impairs neurological function and exacerbates neuropathology after spinal cord injury in rats. J Neurosci 2004; 24: 3752–3761.
Fitch MT, Doller C, Combs CK, Landreth GE, Silver J . Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci 1999; 19: 8182–8198.
Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM . The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol 2002; 61: 623–633.
Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT . Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol 1999; 158: 351–365.
Stirling DP et al. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 2004; 24: 2182–2190.
Wells JE, Hurlbert RJ, Fehlings MG, Yong VW . Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain 2003; 126: 1628–1637.
McPhail LT, Stirling DP, Tetzlaff W, Kwiecien JM, Ramer MS . The contribution of activated phagocytes and myelin degeneration to axonal retraction/dieback following spinal cord injury. Eur J Neurosci 2004; 20: 1984–1994.
Teng YD et al. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci USA 2004; 101: 3071–3076.
Song XY, Zhong JH, Wang X, Zhou XF . Suppression of p75NTR does not promote regeneration of injured spinal cord in mice. J Neurosci 2004; 24: 542–546.
Kwon BK et al. Survival and regeneration of rubrospinal neurons 1 year after spinal cord injury. Proc Natl Acad Sci USA 2002; 99: 3246–3251.
Ruitenberg MJ et al. Adeno-associated viral vector-mediated gene transfer of brain-derived neurotrophic factor reverses atrophy of rubrospinal neurons following both acute and chronic spinal cord injury. Neurobiol Dis 2004; 15: 394–406.
Giehl KM, Tetzlaff W . BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. Eur J Neurosci 1996; 8: 1167–1175.
McDonald JW, Howard MJ . Repairing the damaged spinal cord: a summary of our early success with embryonic stem cell transplantation and remyelination. Prog Brain Res 2002; 137: 299–309.
Kobayashi NR, Fan DP, Giehl KM, Bedard AM, Wiegand SJ, Tetzlaff W . BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Talpha1-tubulin mRNA expression, and promote axonal regeneration. J Neurosci 1997; 17: 9583–9595.
Tobias CA et al. Delayed grafting of BDNF and NT-3 producing fibroblasts into the injured spinal cord stimulates sprouting, partially rescues axotomized red nucleus neurons from loss and atrophy, and provides limited regeneration. Exp Neurol 2003; 184: 97–113.
Lu P, Blesch A, Tuszynski MH . Neurotrophism without neurotropism: BDNF promotes survival but not growth of lesioned corticospinal neurons. J Comp Neurol 2001; 436: 456–470.
Bradbury EJ, King VR, Simmons LJ, Priestley JV, McMahon SB . NT-3, but not BDNF, prevents atrophy and death of axotomized spinal cord projection neurons. Eur J Neurosci 1998; 10: 3058–3068.
Sayer FT, Oudega M, Hagg T . Neurotrophins reduce degeneration of injured ascending sensory and corticospinal motor axons in adult rat spinal cord. Exp Neurol 2002; 175: 282–296.
Cajal SR . Cajal's Degeneration & Regeneration of the Nervous System. Oxford University Press: Oxford 1991.
Caroni P, Schwab ME . Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron 1988; 1: 85–96.
Schnell L, Schwab ME . Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature 1990; 343: 269–272.
Chen MS et al. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 2000; 403: 434–439.
GrandPre T, Nakamura F, Vartanian T, Strittmatter SM . Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 2000; 403: 439–444.
McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE . Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 1994; 13: 805–811.
Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT . A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 1994; 13: 757–767.
Kottis V et al. Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J Neurochem 2002; 82: 1566–1569.
Cai D, Shen Y, De Bellard M, Tang S, Filbin MT . Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron 1999; 22: 89–101.
Bartsch U et al. Lack of evidence that myelin-associated glycoprotein is a major inhibitor of axonal regeneration in the CNS. Neuron 1995; 15: 1375–1381.
Kim JE, Li S, GrandPre T, Qiu D, Strittmatter SM . Axon regeneration in young adult mice lacking Nogo-A/B. Neuron 2003; 38: 187–199.
Simonen M et al. Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron 2003; 38: 201–211.
Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier-Lavigne M . Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron 2003; 38: 213–224.
Huang DW, McKerracher L, Braun PE, David S . A therapeutic vaccine approach to stimulate axon regeneration in the adult mammalian spinal cord. Neuron 1999; 24: 639–647.
Sicotte M, Tsatas O, Jeong SY, Cai CQ, He Z, David S . Immunization with myelin or recombinant Nogo-66/MAG in alum promotes axon regeneration and sprouting after corticospinal tract lesions in the spinal cord. Mol Cell Neurosci 2003; 23: 251–263.
Fournier AE, GrandPre T, Strittmatter SM . Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 2001; 409: 341–346.
Wang KC et al. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 2002; 417: 941–944.
Liu BP, Fournier A, GrandPre T, Strittmatter SM . Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 2002; 297: 1190–1193.
Domeniconi M et al. Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron 2002; 35: 283–290.
GrandPre T, Li S, Strittmatter SM . Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 2002; 417: 547–551.
Li S, Strittmatter SM . Delayed systemic Nogo-66 receptor antagonist promotes recovery from spinal cord injury. J Neurosci 2003; 23: 4219–4227.
MacDermid VE, McPhail LT, Tsang B, Rosenthal A, Davies A, Ramer MS . A soluble form of the Nogo receptor differentially affects plasticity of spinally-projecting axons. Eur J Neurosci 2004; 20: 2567–2579.
Li W et al. A neutralizing anti-Nogo66 receptor monoclonal antibody reverses inhibition of neurite outgrowth by central nervous system myelin. J Biol Chem 2004; 279: 43780–43788.
Wang KC, Kim JA, Sivasankaran R, Segal R, He Z . P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 2002; 420: 74–78.
Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM . A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 2002; 5: 1302–1308.
Mi S et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci 2004; 7: 221–228.
Yamashita T, Tucker KL, Barde YA . Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 1999; 24: 585–593.
Yamashita T, Higuchi H, Tohyama M . The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. J Cell Biol 2002; 157: 565–570.
Yamashita T, Tohyama M . The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci 2003; 6: 461–467.
Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L . Rho signaling pathway targeted to promote spinal cord repair. J Neurosci 2002; 22: 6570–6577.
Fournier AE, Takizawa BT, Strittmatter SM . Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci 2003; 23: 1416–1423.
Monnier PP, Sierra A, Schwab JM, Henke-Fahle S, Mueller BK . The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol Cell Neurosci 2003; 22: 319–330.
Borisoff JF et al. Suppression of Rho-kinase activity promotes axonal growth on inhibitory CNS substrates. Mol Cell Neurosci 2003; 22: 405–416.
Shearer MC et al. The astrocyte/meningeal cell interface is a barrier to neurite outgrowth which can be overcome by manipulation of inhibitory molecules or axonal signalling pathways. Mol Cell Neurosci 2003; 24: 913–925.
Kwon BK, Borisoff JF, Tetzlaff W . Molecular targets for therapeutic intervention after spinal cord injury. Mol Intervent 2002; 2: 244–258.
Keirstead HS, Dyer JK, Sholomenko GN, McGraw J, Delaney KR, Steeves JD . Axonal regeneration and physiological activity following transection and immunological disruption of myelin within the hatchling chick spinal cord. J Neurosci 1995; 15: 6963–6974.
Dyer JK, Bourque JA, Steeves JD . Regeneration of brainstem-spinal axons after lesion and immunological disruption of myelin in adult rat. Exp Neurol 1998; 154: 12–22.
Hiebert GW, Dyer JK, Tetzlaff W, Steeves JD . Immunological myelin disruption does not alter expression of regeneration-associated genes in intact or axotomized rubrospinal neurons. Exp Neurol 2000; 163: 149–156.
Rapalino O et al. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med 1998; 4: 814–821.
Bomstein Y et al. Features of skin-coincubated macrophages that promote recovery from spinal cord injury. J Neuroimmunol 2003; 142: 10–16.
Popovich PG, Jones TB . Manipulating neuroinflammatory reactions in the injured spinal cord: back to basics. Trends Pharmacol Sci 2003; 24: 13–17.
Liu L, Persson JK, Svensson M, Aldskogius H . Glial cell responses, complement, and clusterin in the central nervous system following dorsal root transection. Glia 1998; 23: 221–238.
Ramer MS, Duraisingam I, Priestley JV, McMahon SB . Two-tiered inhibition of axon regeneration at the dorsal root entry zone. J Neurosci 2001; 21: 2651–2660.
Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J . Regeneration of adult axons in white matter tracts of the central nervous system. Nature 1997; 390: 680–683.
Davies SJ, Goucher DR, Doller C, Silver J . Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci 1999; 19: 5810–5822.
Carlstedt T . Regenerating axons form nerve terminals at astrocytes. Brain Res 1985; 347: 188–191.
Zhang Y et al. Correlation between putative inhibitory molecules at the dorsal root entry zone and failure of dorsal root axonal regeneration. Mol Cell Neurosci 2001; 17: 444–459.
Liuzzi FJ, Lasek RJ . Astrocytes block axonal regeneration in mammals by activating the physiological stop pathway. Science 1987; 237: 642–645.
McPhail LT, Plunet W, Das P, Ramer MS . The astrocytic barrier to regeneration at the dorsal root entry zone is induced by rhizotomy. Eur J Neurosci 2005, in press.
Morgenstern DA, Asher RA, Fawcett JW . Chondroitin sulphate proteoglycans in the CNS injury response. Prog Brain Res 2002; 137: 313–332.
Rhodes KE, Fawcett JW . Chondroitin sulphate proteoglycans: preventing plasticity or protecting the CNS? J Anat 2004; 204: 33–48.
Fawcett JW, Asher RA . The glial scar and central nervous system repair. Brain Res Bull 1999; 49: 377–391.
Jones LL, Margolis RU, Tuszynski MH . The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 2003; 182: 399–411.
Tang X, Davies JE, Davies SJ . Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci Res 2003; 71: 427–444.
Properzi F, Asher RA, Fawcett JW . Chondroitin sulphate proteoglycans in the central nervous system: changes and synthesis after injury. Biochem Soc Trans 2003; 31: 335–336.
Schmalfeldt M, Bandtlow CE, Dours-Zimmermann MT, Winterhalter KH, Zimmermann DR . Brain derived versican V2 is a potent inhibitor of axonal growth. J Cell Sci 2000; 113(Part 5): 807–816.
Sugahara K, Mikami T, Uyama T, Mizuguchi S, Nomura K, Kitagawa H . Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol 2003; 13: 612–620.
Dow KE, Ethell DW, Steeves JD, Riopelle RJ . Molecular correlates of spinal cord repair in the embryonic chick: heparan sulfate and chondroitin sulfate proteoglycans. Exp Neurol 1994; 128: 233–238.
McKeon RJ, Hoke A, Silver J . Injury-induced proteoglycans inhibit the potential for laminin-mediated axon growth on astrocytic scars. Exp Neurol 1995; 136: 32–43.
Zuo J, Neubauer D, Dyess K, Ferguson TA, Muir D . Degradation of chondroitin sulfate proteoglycan enhances the neurite-promoting potential of spinal cord tissue. Exp Neurol 1998; 154: 654–662.
Bradbury EJ et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002; 416: 636–640.
Chau CH et al. Chondroitinase ABC enhances axonal regrowth through Schwann cell-seeded guidance channels after spinal cord injury. FASEB J 2004; 18: 194–196.
Moon LD, Asher RA, Rhodes KE, Fawcett JW . Regeneration of CNS axons back to their target following treatment of adult rat brain with chondroitinase ABC. Nat Neurosci 2001; 4: 465–466.
Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L . Reactivation of ocular dominance plasticity in the adult visual cortex. Science 2002; 298: 1248–1251.
Yick LW, Cheung PT, So KF, Wu W . Axonal regeneration of Clarke's neurons beyond the spinal cord injury scar after treatment with chondroitinase ABC. Exp Neurol 2003; 182: 160–168.
Tropea D, Caleo M, Maffei L . Synergistic effects of brain-derived neurotrophic factor and chondroitinase ABC on retinal fiber sprouting after denervation of the superior colliculus in adult rats. J Neurosci 2003; 23: 7034–7044.
Lemons ML, Sandy JD, Anderson DK, Howland DR . Intact aggrecan and chondroitin sulfate-depleted aggrecan core glycoprotein inhibit axon growth in the adult rat spinal cord. Exp Neurol 2003; 184: 981–990.
Grimpe B, Silver J . A novel DNA enzyme reduces glycosaminoglycan chains in the glial scar and allows microtransplanted dorsal root ganglia axons to regenerate beyond lesions in the spinal cord. J Neurosci 2004; 24: 1393–1397.
Iseki K et al. Increased syndecan expression by pleiotrophin and FGF receptor-expressing astrocytes in injured brain tissue. Glia 2002; 39: 1–9.
Garcia-Abreu J, Silva LC, Tovar FF, Onofr GR, Cavalcante LA, Moura NV . Compartmental distribution of sulfated glycosaminoglycans in lateral and medial midbrain astroglial cultures. Glia 1996; 17: 339–344.
Garcia-Abreu J et al. Contribution of heparan sulfate to the non-permissive role of the midline glia to the growth of midbrain neurites. Glia 2000; 29: 260–272.
Cavalcante LA, Garcia-Abreu J, Moura NV, Silva LC, Weissmuller G . Modulators of axonal growth and guidance at the brain midline with special reference to glial heparan sulfate proteoglycans. An Acad Bras Cienc 2002; 74: 691–716.
Weissmuller G, Garcia-Abreu J, Mascarello BP, Moura NV, Cavalcante LA . Glial cells with differential neurite growth-modulating properties probed by atomic force microscopy. Neurosci Res 2000; 38: 217–220.
Walz A, McFarlane S, Brickman YG, Nurcombe V, Bartlett PF, Holt CE . Essential role of heparan sulfates in axon navigation and targeting in the developing visual system. Development 1997; 124: 2421–2430.
Carey DJ . Syndecans: multifunctional cell-surface co-receptors. Biochem J 1997; 327(Part 1): 1–16.
Vijayan VK, Lee YL, Eng LF . Immunohistochemical localization of basic fibroblast growth factor in cultured rat astrocytes and oligodendrocytes. Int J Dev Neurosci 1993; 11: 257–267.
Eclancher F, Kehrli P, Labourdette G, Sensenbrenner M . Basic fibroblast growth factor (bFGF) injection activates the glial reaction in the injured adult rat brain. Brain Res 1996; 737: 201–214.
Bansal R, Kumar M, Murray K, Pfeiffer SE . Developmental and FGF-2-mediated regulation of syndecans (1–4) and glypican in oligodendrocytes. Mol Cell Neurosci 1996; 7: 276–288.
Karthikeyan L, Flad M, Engel M, Meyer-Puttlitz B, Margolis RU, Margolis RK . Immunocytochemical and in situ hybridization studies of the heparan sulfate proteoglycan, glypican, in nervous tissue. J Cell Sci 1994; 107(Part 11): 3213–3222.
Litwack ED, Stipp CS, Kumbasar A, Lander AD . Neuronal expression of glypican, a cell-surface glycosylphosphatidylinositol-anchored heparan sulfate proteoglycan, in the adult rat nervous system. J Neurosci 1994; 14: 3713–3724.
Funderburgh JL et al. Sequence, molecular properties, and chromosomal mapping of mouse lumican. Invest Ophthalmol Vis Sci 1995; 36: 2296–2303.
Funderburgh JL . Keratan sulfate: structure, biosynthesis, and function. Glycobiology 2000; 10: 951–958.
Bareyre FM, Haudenschild B, Schwab ME . Long-lasting sprouting and gene expression changes induced by the monoclonal antibody IN-1 in the adult spinal cord. J Neurosci 2002; 22: 7097–7110.
Seo H, Geisert Jr EE . A keratan sulfate proteoglycan marks the boundaries in the cortical barrel fields of the adult rat. Neurosci Lett 1995; 197: 13–16.
Cole GJ, McCabe CF . Identification of a developmentally regulated keratan sulfate proteoglycan that inhibits cell adhesion and neurite outgrowth. Neuron 1991; 7: 1007–1018.
McCabe CF, Cole GJ . Expression of the barrier-associated proteins EAP-300 and claustrin in the developing central nervous system. Brain Res Dev Brain Res 1992; 70: 9–24.
Geisert Jr EE, Bidanset DJ . A central nervous system keratan sulfate proteoglycan: localization to boundaries in the neonatal rat brain. Brain Res Dev Brain Res 1993; 75: 163–173.
Robson JA, Geisert Jr EE . Expression of a keratin sulfate proteoglycan during development of the dorsal lateral geniculate nucleus in the ferret. J Comp Neurol 1994; 340: 349–360.
Jones LL, Tuszynski MH . Spinal cord injury elicits expression of keratan sulfate proteoglycans by macrophages, reactive microglia, and oligodendrocyte progenitors. J Neurosci 2002; 22: 4611–4624.
Miller B, Sheppard AM, Pearlman AL . Developmental expression of keratan sulfate-like immunoreactivity distinguishes thalamic nuclei and cortical domains. J Comp Neurol 1997; 380: 533–552.
Gilad GM, Kagan HM, Gilad VH . Lysyl oxidase, the extracellular matrix-forming enzyme, in rat brain injury sites. Neurosci Lett 2001; 310: 45–48.
Kagan HM, Trackman PC . Properties and function of lysyl oxidase. Am J Respir Cell Mol Biol 1991; 5: 206–210.
Kagan HM, Li W . Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem 2003; 88: 660–672.
Ellison JA et al. Osteopontin and its integrin receptor alpha(v)beta3 are upregulated during formation of the glial scar after focal stroke. Stroke 1998; 29: 1698–1706.
Wang X et al. Delayed expression of osteopontin after focal stroke in the rat. J Neurosci 1998; 18: 2075–2083.
Fu Y, Hashimoto M, Ino H, Murakami M, Yamazaki M, Moriya H . Spinal root avulsion-induced upregulation of osteopontin expression in the adult rat spinal cord. Acta Neuropathol (Berl) 2004; 107: 8–16.
Ellison JA, Barone FC, Feuerstein GZ . Matrix remodeling after stroke. De novo expression of matrix proteins and integrin receptors. Ann NY Acad Sci 1999; 890: 204–222.
Pindon A, Berry M, Hantai D . Thrombomodulin as a new marker of lesion-induced astrogliosis: involvement of thrombin through the G-protein-coupled protease-activated receptor-1. J Neurosci 2000; 20: 2543–2550.
Jaworski DM . Differential regulation of tissue inhibitor of metalloproteinase mRNA expression in response to intracranial injury. Glia 2000; 30: 199–208.
Muir EM et al. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res Mol Brain Res 2002; 100: 103–117.
Mori T, Iijima N, Kitabatake K, Kohsaka S . Alpha 2-macroglobulin is an astroglia-derived neurite-promoting factor for cultured neurons from rat central nervous system. Brain Res 1990; 527: 55–61.
Logan A, Baird A, Berry M . Decorin attenuates gliotic scar formation in the rat cerebral hemisphere. Exp Neurol 1999; 159: 504–510.
Mendis DB, Ivy GO, Brown IR . SC1, a brain extracellular matrix glycoprotein related to SPARC and follistatin, is expressed by rat cerebellar astrocytes following injury and during development. Brain Res 1996; 730: 95–106.
Mendis DB, Ivy GO, Brown IR . Induction of SC1 mRNA encoding a brain extracellular matrix glycoprotein related to SPARC following lesioning of the adult rat forebrain. Neurochem Res 2000; 25: 1637–1644.
Richardson PM, Issa VM . Peripheral injury enhances central regeneration of primary sensory neurones. Nature 1984; 309: 791–793.
Chong MS, Woolf CJ, Haque NS, Anderson PN . Axonal regeneration from injured dorsal roots into the spinal cord of adult rats. J Comp Neurol 1999; 410: 42–54.
Neumann S, Woolf CJ . Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 1999; 23: 83–91.
Bulsara KR, Iskandar BJ, Villavicencio AT, Skene JH . A new millenium for spinal cord regeneration: growth-associated genes. Spine 2002; 27: 1946–1949.
Qiu J, Cai D, Filbin MT . A role for cAMP in regeneration during development and after injury. Prog Brain Res 2002; 137: 381–387.
Chaisuksunt V et al. Axonal regeneration from CNS neurons in the cerebellum and brainstem of adult rats: correlation with the patterns of expression and distribution of messenger RNAs for L1, CHL1, c-jun and growth-associated protein-43. Neuroscience 2000; 100: 87–108.
Chong MS et al. GAP-43 expression in primary sensory neurons following central axotomy. J Neurosci 1994; 14: 4375–4384.
Ramer MS, Priestley JV, McMahon SB . Functional regeneration of sensory axons into the adult spinal cord. Nature 2000; 403: 312–316.
Ramer MS et al. Neurotrophin-3-mediated regeneration and recovery of proprioception following dorsal rhizotomy. Mol Cell Neurosci 2002; 19: 239–249.
Qiu J et al. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 2002; 34: 895–903.
Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT . Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci 2001; 21: 4731–4739.
Cai D, Deng K, Mellado W, Lee J, Ratan RR, Filbin MT . Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 2002; 35: 711–719.
Hebenstreit GF et al. Rolipram in major depressive disorder: results of a double-blind comparative study with imipramine. Pharmacopsychiatry 1989; 22: 156–160.
Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT . The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci USA 2004; 101: 8786–8790.
Pearse DD et al. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med 2004; 10: 610–616.
Houle JD, Johnson JE . Nerve growth factor (NGF)-treated nitrocellulose enhances and directs the regeneration of adult rat dorsal root axons through intraspinal neural tissue transplants. Neurosci Lett 1989; 103: 17–23.
Chuah MI, West AK . Cellular and molecular biology of ensheathing cells. Microsc Res Tech 2002; 58: 216–227.
Oudega M, Hagg T . Neurotrophins promote regeneration of sensory axons in the adult rat spinal cord. Brain Res 1999; 818: 431–438.
Xu XM, Guenard V, Kleitman N, Aebischer P, Bunge MB . A combination of BDNF and NT-3 promotes supraspinal axonal regeneration into Schwann cell grafts in adult rat thoracic spinal cord. Exp Neurol 1995; 134: 261–272.
Bregman BS, McAtee M, Dai HN, Kuhn PL . Neurotrophic factors increase axonal growth after spinal cord injury and transplantation in the adult rat. Exp Neurol 1997; 148: 475–494.
Lu P, Yang H, Jones LL, Filbin MT, Tuszynski MH . Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J Neurosci 2004; 24: 6402–6409.
Bradbury EJ, Khemani S, Von R, King, Priestley JV, McMahon SB . NT-3 promotes growth of lesioned adult rat sensory axons ascending in the dorsal columns of the spinal cord. Eur J Neurosci 1999; 11: 3873–3883.
Hendriks WT, Ruitenberg MJ, Blits B, Boer GJ, Verhaagen J . Viral vector-mediated gene transfer of neurotrophins to promote regeneration of the injured spinal cord. Prog Brain Res 2004; 146: 451–476.
Ramer MS, McMahon SB, Priestley JV . Axon regeneration across the dorsal root entry zone. Prog Brain Res 2001; 132: 621–639.
Zhang Y, Dijkhuizen PA, Anderson PN, Lieberman AR, Verhaagen J . NT-3 delivered by an adenoviral vector induces injured dorsal root axons to regenerate into the spinal cord of adult rats. J Neurosci Res 1998; 54: 554–562.
Bennett DL et al. A distinct subgroup of small DRG cells express GDNF receptor components and GDNF is protective for these neurons after nerve injury. J Neurosci 1998; 18: 3059–3072.
Iwakawa M, Mizoi K, Tessler A, Itoh Y . Intraspinal implants of fibrin glue containing glial cell line-derived neurotrophic factor promote dorsal root regeneration into spinal cord. Neurorehabil Neural Repair 2001; 15: 173–182.
Romero MI, Rangappa N, Li L, Lightfoot E, Garry MG, Smith GM . Extensive sprouting of sensory afferents and hyperalgesia induced by conditional expression of nerve growth factor in the adult spinal cord. J Neurosci 2000; 20: 4435–4445.
Romero MI, Rangappa N, Garry MG, Smith GM . Functional regeneration of chronically injured sensory afferents into adult spinal cord after neurotrophin gene therapy. J Neurosci 2001; 21: 8408–8416.
Ramer MS, Bradbury EJ, McMahon SB . Nerve growth factor induces P2X(3) expression in sensory neurons. J Neurochem 2001; 77: 864–875.
Tang XQ, Tanelian DL, Smith GM . Semaphorin3A inhibits nerve growth factor-induced sprouting of nociceptive afferents in adult rat spinal cord. J Neurosci 2004; 24: 819–827.
Krenz NR, Meakin SO, Krassioukov AV, Weaver LC . Neutralizing intraspinal nerve growth factor blocks autonomic dysreflexia caused by spinal cord injury. J Neurosci 1999; 19: 7405–7414.
Marsh DR, Wong ST, Meakin SO, MacDonald JI, Hamilton EF, Weaver LC . Neutralizing intraspinal nerve growth factor with a trkA-IgG fusion protein blocks the development of autonomic dysreflexia in a clip-compression model of spinal cord injury. J Neurotrauma 2002; 19: 1531–1541.
Hempstead BL . The many faces of p75NTR. Curr Opin Neurobiol 2002; 12: 260–267.
MacPhee IJ, Barker PA . Brain-derived neurotrophic factor binding to the p75 neurotrophin receptor reduces TrkA signaling while increasing serine phosphorylation in the TrkA intracellular domain. J Biol Chem 1997; 272: 23547–23551.
Kimpinski K, Jelinski S, Mearow K . The anti-p75 antibody, MC192, and brain-derived neurotrophic factor inhibit nerve growth factor-dependent neurite growth from adult sensory neurons. Neuroscience 1999; 93: 253–263.
Kohn J, Aloyz RS, Toma JG, Haak-Frendscho M, Miller FD . Functionally antagonistic interactions between the TrkA and p75 neurotrophin receptors regulate sympathetic neuron growth and target innervation. J Neurosci 1999; 19: 5393–5408.
Yeo TT et al. Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci 1997; 17: 7594–7605.
Ferri CC, Moore FA, Bisby MA . Effects of facial nerve injury on mouse motoneurons lacking the p75 low-affinity neurotrophin receptor. J Neurobiol 1998; 34: 1–9.
Boyd JG, Gordon T . The neurotrophin receptors, trkB and p75, differentially regulate motor axonal regeneration. J Neurobiol 2001; 49: 314–325.
Zhou XF et al. Satellite-cell-derived nerve growth factor and neurotrophin-3 are involved in noradrenergic sprouting in the dorsal root ganglia following peripheral nerve injury in the rat. Eur J Neurosci 1999; 11: 1711–1722.
Ramer MS, Thompson SW, McMahon SB . Causes and consequences of sympathetic basket formation in dorsal root ganglia. Pain 1999; (Suppl 6): S111–S120.
Walsh GS, Krol KM, Kawaja MD . Absence of the p75 neurotrophin receptor alters the pattern of sympathosensory sprouting in the trigeminal ganglia of mice overexpressing nerve growth factor. J Neurosci 1999; 19: 258–273.
Scott ALM, Borisoff JF, Ramer MS . Deafferentation and neurotrophin-mediated intraspinal sprouting: a central role for the p75 neurotrophin receptor. Eur J Neurosci 2004, in press.
Oudega M, Rosano C, Sadi D, Wood PM, Schwab ME, Hagg T . Neutralizing antibodies against neurite growth inhibitor NI-35/250 do not promote regeneration of sensory axons in the adult rat spinal cord. Neuroscience 2000; 100: 873–883.
Goldberger ME . Locomotor recovery after unilateral hindlimb deafferentation in cats. Brain Res 1977; 123: 59–74.
Ballermann M, McKenna J, Whishaw IQ . A grasp-related deficit in tactile discrimination following dorsal column lesion in the rat. Brain Res Bull 2001; 54: 237–242.
Liu CN, Chambers WW . Intraspinal sprouting of dorsal root axons. Archs Neurol Psychiatr 1998; 79: 46–61.
Jeffery ND, Fitzgerald M . Effects of red nucleus ablation and exogenous neurotrophin-3 on corticospinal axon terminal distribution in the adult rat. Neuroscience 2001; 104: 513–521.
Illis LS . Experimental model of regeneration in the central nervous system. II. The reaction of glia in the synaptic zone. Brain 1973; 96: 61–68.
Illis L . Spinal cord synapses in the cat: the reaction of the boutons terminaux at the motoneurone surface to experimental denervation. Brain 1964; 87: 555–572.
Illis L . Changes in spinal cord synapses and a possible explanation for spinal shock. Exp Neurol 1963; 8: 328–335.
Illis LS . Experimental model of regeneration in the central nervous system. I. Synaptic changes. Brain 1973; 96: 47–60.
Illis L . Spinal cord synapses in the cat: the normal appearances by light microscope. Brain 1964; 87: 543–554.
Muir GD, Steeves JD . Sensorimotor stimulation to improve locomotor recovery after spinal cord injury. Trends Neurosci 1997; 20: 72–77.
Dunlop SA, Steeves JD . Neural activity and facilitated recovery by training after CNS injury: implications for rehabilitation. Top Spinal Cord Inj Rehabil 2003; 8: 92–103.
Llewellyn-Smith IJ, Weaver LC . Changes in synaptic inputs to sympathetic preganglionic neurons after spinal cord injury. J Comp Neurol 2001; 435: 226–240.
Belyantseva IA, Lewin GR . Stability and plasticity of primary afferent projections following nerve regeneration and central degeneration. Eur J Neurosci 1999; 11: 457–468.
Wang SD, Goldberger ME, Murray M . Plasticity of spinal systems after unilateral lumbosacral dorsal rhizotomy in the adult rat. J Comp Neurol 1991; 304: 555–568.
Carlstedt T . Dorsal root innervation of spinal cord neurons after dorsal root implantation into the spinal cord of adult rats. Neurosci Lett 1985; 55: 343–348.
Smith KJ, Kodama RT, Liuzzi FJ . Is CNS trauma a prerequisite for the elongation of CNS axons into denervated peripheral nerve? Brain Res 1992; 575: 79–85.
Schnell L, Schneider R, Kolbeck R, Barde YA, Schwab ME . Neurotrophin-3 enhances sprouting of corticospinal tract during development and after adult spinal cord lesion. Nature 1994; 367: 170–173.
Thallmair M, Metz GA, Z'Graggen WJ, Raineteau O, Kartje GL, Schwab ME . Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nat Neurosci 1998; 1: 124–131.
Richardson PM, McGuinness UM, Aguayo AJ . Axons from CNS neurons regenerate into PNS grafts. Nature 1980; 284: 264–265.
Cheng H, Cao Y, Olson L . Spinal cord repair in adult paraplegic rats: partial restoration of hind limb function. Science 1996; 273: 510–513.
Lee YS, Hsiao I, Lin VW . Peripheral nerve grafts and aFGF restore partial hindlimb function in adult paraplegic rats. J Neurotrauma 2002; 19: 1203–1216.
Fraidakis MJ, Spenger C, Olson L . Partial recovery after treatment of chronic paraplegia in rat. Exp Neurol 2004; 188: 33–42.
Cheng H, Liao KK, Liao SF, Chuang TY, Shih YH . Spinal cord repair with acidic fibroblast growth factor as a treatment for a patient with chronic paraplegia. Spine 2004; 29: E284–E288.
Steeves J, Fawcett J, Tuszynski M . Report of international clinical trials workshop on spinal cord injury february 20–21, 2004, vancouver, Canada. Spinal Cord 2004; 42: 591–597.
Tadie M et al. Partial return of motor function in paralyzed legs after surgical bypass of the lesion site by nerve autografts three years after spinal cord injury. J Neurotrauma 2002; 19: 909–916.
Hall SM . The effect of inhibiting Schwann cell mitosis on the re-innervation of acellular autografts in the peripheral nervous system of the mouse. Neuropathol Appl Neurobiol 1986; 12: 401–414.
Hall SM . Regeneration in cellular and acellular autografts in the peripheral nervous system. Neuropathol Appl Neurobiol 1986; 12: 27–46.
Fawcett JW, Keynes RJ . Peripheral nerve regeneration. Annu Rev Neurosci 1990; 13: 43–60.
Mirsky R et al. Schwann cells as regulators of nerve development. J Physiol Paris 2002; 96: 17–24.
Casella GT, Bunge RP, Wood PM . Improved method for harvesting human Schwann cells from mature peripheral nerve and expansion in vitro. Glia 1996; 17: 327–338.
Guest JD, Hesse D, Schnell L, Schwab ME, Bunge MB, Bunge RP . Influence of IN-1 antibody and acidic FGF-fibrin glue on the response of injured corticospinal tract axons to human Schwann cell grafts. J Neurosci Res 1997; 50: 888–905.
Guest JD, Rao A, Olson L, Bunge MB, Bunge RP . The ability of human Schwann cell grafts to promote regeneration in the transected nude rat spinal cord. Exp Neurol 1997; 148: 502–522.
Bunge MB . Bridging the transected or contused adult rat spinal cord with Schwann cell and olfactory ensheathing glia transplants. Prog Brain Res 2002; 137: 275–282.
Xu XM, Guenard V, Kleitman N, Bunge MB . Axonal regeneration into Schwann cell-seeded guidance channels grafted into transected adult rat spinal cord. J Comp Neurol 1995; 351: 145–160.
Xu XM, Chen A, Guenard V, Kleitman N, Bunge MB . Bridging Schwann cell transplants promote axonal regeneration from both the rostral and caudal stumps of transected adult rat spinal cord. J Neurocytol 1997; 26: 1–16.
Xu XM, Zhang SX, Li H, Aebischer P, Bunge MB . Regrowth of axons into the distal spinal cord through a Schwann-cell-seeded mini-channel implanted into hemisected adult rat spinal cord. Eur J Neurosci 1999; 11: 1723–1740.
Bunge MB . Transplantation of purified populations of Schwann cells into lesioned adult rat spinal cord. J Neurol 1994; 242: S36–S39.
Chen A, Xu XM, Kleitman N, Bunge MB . Methylprednisolone administration improves axonal regeneration into Schwann cell grafts in transected adult rat thoracic spinal cord. Exp Neurol 1996; 138: 261–276.
Pinzon A, Calancie B, Oudega M, Noga BR . Conduction of impulses by axons regenerated in a Schwann cell graft in the transected adult rat thoracic spinal cord. J Neurosci Res 2001; 64: 533–541.
Plant GW, Bates ML, Bunge MB . Inhibitory proteoglycan immunoreactivity is higher at the caudal than the rostral Schwann cell graft-transected spinal cord interface. Mol Cell Neurosci 2001; 17: 471–487.
Graziadei PP, Monti Graziadei GA . Neurogenesis and neuron regeneration in the olfactory system of mammals. III. Deafferentation and reinnervation of the olfactory bulb following section of the fila olfactoria in rat. J Neurocytol 1980; 9: 145–162.
Doucette JR, Kiernan JA, Flumerfelt BA . The re-innervation of olfactory glomeruli following transection of primary olfactory axons in the central or peripheral nervous system. J Anat 1983; 137(Part 1): 1–19.
Deckner ML, Risling M, Frisen J . Apoptotic death of olfactory sensory neurons in the adult rat. Exp Neurol 1997; 143: 132–140.
Harding J, Graziadei PP, Monti Graziadei GA, Margolis FL . Denervation in the primary olfactory pathway of mice. IV. Biochemical and morphological evidence for neuronal replacement following nerve section. Brain Res 1977; 132: 11–28.
Kern RC, Quinn B, Rosseau G, Farbman AI . Post-traumatic olfactory dysfunction. Laryngoscope 2000; 110: 2106–2109.
Ramon-Cueto A, Nieto-Sampedro M . Regeneration into the spinal cord of transected dorsal root axons is promoted by ensheathing glia transplants. Exp Neurol 1994; 127: 232–244.
Navarro X et al. Ensheathing glia transplants promote dorsal root regeneration and spinal reflex restitution after multiple lumbar rhizotomy. Ann Neurol 1999; 45: 207–215.
Taylor JS, Muneton-Gomez VC, Eguia-Recuero R, Nieto-Sampedro M . Transplants of olfactory bulb ensheathing cells promote functional repair of multiple dorsal rhizotomy. Prog Brain Res 2001; 132: 641–654.
Pascual JI, Gudino-Cabrera G, Insausti R, Nieto-Sampedro M . Spinal implants of olfactory ensheathing cells promote axon regeneration and bladder activity after bilateral lumbosacral dorsal rhizotomy in the adult rat. J Urol 2002; 167: 1522–1526.
Gomez VM et al. Transplantation of olfactory ensheathing cells fails to promote significant axonal regeneration from dorsal roots into the rat cervical cord. J Neurocytol 2003; 32: 53–70.
Riddell JS, Enriquez-Denton M, Toft A, Fairless R, Barnett SC . Olfactory ensheathing cell grafts have minimal influence on regeneration at the dorsal root entry zone following rhizotomy. Glia 2004; 47: 150–167.
Ramer LM, Richter MW, Roskams AJ, Tetzlaff W, Ramer MS . Peripherally-derived olfactory ensheathing cells do not promote primary afferent regeneration following dorsal root injury. Glia 2004; 47: 189–206.
Ramon-Cueto A, Plant GW, Avila J, Bunge MB . Long-distance axonal regeneration in the transected adult rat spinal cord is promoted by olfactory ensheathing glia transplants. J Neurosci 1998; 18: 3803–3815.
Li Y, Field PM, Raisman G . Repair of adult rat corticospinal tract by transplants of olfactory ensheathing cells. Science 1997; 277: 2000–2002.
Li Y, Field PM, Raisman G . Regeneration of adult rat corticospinal axons induced by transplanted olfactory ensheathing cells. J Neurosci 1998; 18: 10514–10524.
Nash HH, Borke RC, Anders JJ . Ensheathing cells and methylprednisolone promote axonal regeneration and functional recovery in the lesioned adult rat spinal cord. J Neurosci 2002; 22: 7111–7120.
Ramon-Cueto A, Cordero MI, Santos-Benito FF, Avila J . Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron 2000; 25: 425–435.
Lu J, Feron F, Ho SM, Mackay-Sim A, Waite PM . Transplantation of nasal olfactory tissue promotes partial recovery in paraplegic adult rats. Brain Res 2001; 889: 344–357.
Huang H et al. Influence of patients' age on functional recovery after transplantation of olfactory ensheathing cells into injured spinal cord injury. Chin Med J (Engl) 2003; 116: 1488–1491.
Bianco JI, Perry C, Harkin DG, Mackay-Sim A, Feron F . Neurotrophin 3 promotes purification and proliferation of olfactory ensheathing cells from human nose. Glia 2004; 45: 111–123.
Verdu E, Garcia-Alias G, Fores J, Lopez-Vales R, Navarro X . Olfactory ensheathing cells transplanted in lesioned spinal cord prevent loss of spinal cord parenchyma and promote functional recovery. Glia 2003; 42: 275–286.
Verdu E et al. Effects of ensheathing cells transplanted into photochemically damaged spinal cord. Neuroreport 2001; 12: 2303–2309.
Takami T, Oudega M, Bates ML, Wood PM, Kleitman N, Bunge MB . Schwann cell but not olfactory ensheathing glia transplants improve hindlimb locomotor performance in the moderately contused adult rat thoracic spinal cord. J Neurosci 2002; 22: 6670–6681.
Plant GW, Christensen CL, Oudega M, Bunge MB . Delayed transplantation of olfactory ensheathing glia promotes sparing/regeneration of supraspinal axons in the contused adult rat spinal cord. J Neurotrauma 2003; 20: 1–16.
Ruitenberg MJ et al. Ex vivo adenoviral vector-mediated neurotrophin gene transfer to olfactory ensheathing glia: effects on rubrospinal tract regeneration, lesion size, and functional recovery after implantation in the injured rat spinal cord. J Neurosci 2003; 23: 7045–7058.
Ramer LM, Au E, Richter MW, Liu J, Tetzlaff W, Roskams AJ . Peripheral olfactory ensheathing cells reduce scar and cavity formation and promote regeneration after spinal cord injury. J Comp Neurol 2004; 473: 1–15.
Chuah MI et al. Olfactory ensheathing cells promote collateral axonal branching in the injured adult rat spinal cord. Exp Neurol 2004; 185: 15–25.
Barnett SC, Chang L . Olfactory ensheathing cells and CNS repair: going solo or in need of a friend? Trends Neurosci 2004; 27: 54–60.
Li Y, Decherchi P, Raisman G . Transplantation of olfactory ensheathing cells into spinal cord lesions restores breathing and climbing. J Neurosci 2003; 23: 727–731.
Lu J, Feron F, Mackay-Sim A, Waite PM . Olfactory ensheathing cells promote locomotor recovery after delayed transplantation into transected spinal cord. Brain 2002; 125: 14–21.
Keyvan-Fouladi N, Raisman G, Li Y . Functional repair of the corticospinal tract by delayed transplantation of olfactory ensheathing cells in adult rats. J Neurosci 2003; 23: 9428–9434.
Wewetzer K, Verdu E, Angelov DN, Navarro X . Olfactory ensheathing glia and Schwann cells: two of a kind? Cell Tissue Res 2002; 309: 337–345.
Williams SK, Franklin RJ, Barnett SC . Response of olfactory ensheathing cells to the degeneration and regeneration of the peripheral olfactory system and the involvement of the neuregulins. J Comp Neurol 2004; 470: 50–62.
Menei P, Montero-Menei C, Whittemore SR, Bunge RP, Bunge MB . Schwann cells genetically modified to secrete human BDNF promote enhanced axonal regrowth across transected adult rat spinal cord. Eur J Neurosci 1998; 10: 607–621.
Cao L et al. Olfactory ensheathing cells genetically modified to secrete GDNF to promote spinal cord repair. Brain 2004; 127: 535–549.
Lakatos A, Franklin RJ, Barnett SC . Olfactory ensheathing cells and Schwann cells differ in their in vitro interactions with astrocytes. Glia 2000; 32: 214–225.
Lakatos A, Barnett SC, Franklin RJ . Olfactory ensheathing cells induce less host astrocyte response and chondroitin sulphate proteoglycan expression than Schwann cells following transplantation into adult CNS white matter. Exp Neurol 2003; 184: 237–246.
Woodhall E, West AK, Vickers JC, Chuah MI . Olfactory ensheathing cell phenotype following implantation in the lesioned spinal cord. Cell Mol Life Sci 2003; 60: 2241–2253.
Cui Q, Pollett MA, Symons NA, Plant GW, Harvey AR . A new approach to CNS repair using chimeric peripheral nerve grafts. J Neurotrauma 2003; 20: 17–31.
Pearse DD, Marcillo AE, Oudega M, Lynch MP, Wood PM, Bunge MB . Transplantation of Schwann cells and olfactory ensheathing glia after spinal cord injury: does pretreatment with methylprednisolone and interleukin-10 enhance recovery? J Neurotrauma 2004; 21: 1223–1239.
Boyd JG, Lee J, Skihar V, Doucette R, Kawaja MD . LacZ-expressing olfactory ensheathing cells do not associate with myelinated axons after implantation into the compressed spinal cord. Proc Natl Acad Sci USA 2004; 101: 2162–2166.
Sims TJ, Gilmore SA . Regrowth of dorsal root axons into a radiation-induced glial-deficient environment in the spinal cord. Brain Res 1994; 634: 113–126.
Sims TJ, Gilmore SA . Regeneration of dorsal root axons into experimentally altered glial environments in the rat spinal cord. Exp Brain Res 1994; 99: 25–33.
Ramer MS . Spontaneous functional viscerosensory regeneration into the adult brainstem. J Neurosci 2003; 23: 9770–9775.
Shimizu I, Oppenheim RW, O'Brien M, Shneiderman A . Anatomical and functional recovery following spinal cord transection in the chick embryo. J Neurobiol 1990; 21: 918–937.
Hasan SJ, Keirstead HS, Muir GD, Steeves JD . Axonal regeneration contributes to repair of injured brainstem–spinal neurons in embryonic chick. J Neurosci 1993; 13: 492–507.
Varga ZM, Schwab ME, Nicholls JG . Myelin-associated neurite growth-inhibitory proteins and suppression of regeneration of immature mammalian spinal cord in culture. Proc Natl Acad Sci USA 1995; 92: 10959–10963.
Steeves JD . Cues from developmental models of spinal cord regeneration for the repair of the injured adult CNS. In: Ingoglia NA, Murray M (eds). Axonal Regeneration in the Central Nervous System. Marcel Dekker: Basel 2000 pp 129–159.
Bregman BS, Kunkel-Bagden E, Reier PJ, Dai HN, McAtee M, Gao D . Recovery of function after spinal cord injury: mechanisms underlying transplant-mediated recovery of function differ after spinal cord injury in newborn and adult rats. Exp Neurol 1993; 123: 3–16.
Houle JD, Reier PJ . Transplantation of fetal spinal cord tissue into the chronically injured adult rat spinal cord. J Comp Neurol 1988; 269: 535–547.
Zurita M, Vaquero J, Oya S, Montilla J . Functional recovery in chronic paraplegic rats after co-grafts of fetal brain and adult peripheral nerve tissue. Surg Neurol 2001; 55: 249–254.
Anderson DK, Howland DR, Reier PJ . Fetal neural grafts and repair of the injured spinal cord. Brain Pathol 1995; 5: 451–457.
Lakatos A, Franklin RJ . Transplant mediated repair of the central nervous system: an imminent solution? Curr Opin Neurol 2002; 15: 701–705.
Jakeman LB, Reier PJ . Axonal projections between fetal spinal cord transplants and the adult rat spinal cord: a neuroanatomical tracing study of local interactions. J Comp Neurol 1991; 307: 311–334.
Duchossoy Y, Kassar-Duchossoy L, Orsal D, Stettler O, Horvat JC . Reinnervation of the biceps brachii muscle following cotransplantation of fetal spinal cord and autologous peripheral nerve into the injured cervical spinal cord of the adult rat. Exp Neurol 2001; 167: 329–340.
Coumans JV et al. Axonal regeneration and functional recovery after complete spinal cord transection in rats by delayed treatment with transplants and neurotrophins. J Neurosci 2001; 21: 9334–9344.
Wirth III ED et al. Feasibility and safety of neural tissue transplantation in patients with syringomyelia. J Neurotrauma 2001; 18: 911–929.
Thompson FJ et al. Neurophysiological assessment of the feasibility and safety of neural tissue transplantation in patients with syringomyelia. J Neurotrauma 2001; 18: 931–945.
Chow SY et al. Characterization and intraspinal grafting of EGF/bFGF-dependent neurospheres derived from embryonic rat spinal cord. Brain Res 2000; 874: 87–106.
Lu P, Jones LL, Snyder EY, Tuszynski MH . Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp Neurol 2003; 181: 115–129.
Roy NS et al. Telomerase immortalization of neuronally restricted progenitor cells derived from the human fetal spinal cord. Nat Biotechnol 2004; 22: 297–305.
Ogawa Y et al. Transplantation of in vitro-expanded fetal neural progenitor cells results in neurogenesis and functional recovery after spinal cord contusion injury in adult rats. J Neurosci Res 2002; 69: 925–933.
Cao Q, Benton RL, Whittemore SR . Stem cell repair of central nervous system injury. J Neurosci Res 2002; 68: 501–510.
Cao QL, Zhang YP, Howard RM, Walters WM, Tsoulfas P, Whittemore SR . Pluripotent stem cells engrafted into the normal or lesioned adult rat spinal cord are restricted to a glial lineage. Exp Neurol 2001; 167: 48–58.
Teng YD et al. Functional recovery following traumatic spinal cord injury mediated by a unique polymer scaffold seeded with neural stem cells. Proc Natl Acad Sci USA 2002; 99: 3024–3029.
Rabinovich SS et al. Transplantation treatment of spinal cord injury patients. Biomed Pharmacother 2003; 57: 428–433.
Calza L et al. Stem cells and nervous tissue repair: from in vitro to in vivo. Prog Brain Res 2004; 146: 75–91.
Rothstein JD, Snyder EY . Reality and immortality – neural stem cells for therapies. Nat Biotechnol 2004; 22: 283–285.
Jimenez Hamann MC, Tsai EC, Tator CH, Shoichet MS . Novel intrathecal delivery system for treatment of spinal cord injury. Exp Neurol 2003; 182: 300–309.
Cao X, Shoichet MS . Investigating the synergistic effect of combined neurotrophic factor concentration gradients to guide axonal growth. Neuroscience 2003; 122: 381–389.
Kapur TA, Shoichet MS . Immobilized concentration gradients of nerve growth factor guide neurite outgrowth. J Biomed Mater Res 2004; 68A: 235–243.
Patist CM, Mulder MB, Gautier SE, Maquet V, Jerome R, Oudega M . Freeze-dried poly(D,L-lactic acid) macroporous guidance scaffolds impregnated with brain-derived neurotrophic factor in the transected adult rat thoracic spinal cord. Biomaterials 2004; 25: 1569–1582.
Tsai EC, Dalton PD, Shoichet MS, Tator CH . Synthetic hydrogel guidance channels facilitate regeneration of adult rat brainstem motor axons after complete spinal cord transection. J Neurotrauma 2004; 21: 789–804.
Geller HM, Fawcett JW . Building a bridge: engineering spinal cord repair. Exp Neurol 2002; 174: 125–136.
Friedman JA, Windebank AJ, Moore MJ, Spinner RJ, Currier BL, Yaszemski MJ . Biodegradable polymer grafts for surgical repair of the injured spinal cord. Neurosurgery 2002; 51: 742–751.
Novikova LN, Novikov LN, Kellerth JO . Biopolymers and biodegradable smart implants for tissue regeneration after spinal cord injury. Curr Opin Neurol 2003; 16: 711–715.
Smith EJ, Blakemore WF, McDonald WI . Central remyelination restores secure conduction. Nature 1979; 280: 395–396.
Jeffery ND, Blakemore WF . Locomotor deficits induced by experimental spinal cord demyelination are abolished by spontaneous remyelination. Brain 1997; 120(Part 1): 27–37.
Bunge RP . Expanding roles for the Schwann cell: ensheathment, myelination, trophism and regeneration. Curr Opin Neurobiol 1993; 3: 805–809.
Blakemore WF, Patterson RC . Observations on the interactions of Schwann cells and astrocytes following X-irradiation of neonatal rat spinal cord. J Neurocytol 1975; 4: 573–585.
Felts PA, Smith KJ, Tilt E . Blood–brain barrier function in central demyelinating lesions repaired by Schwann cell remyelination. Ann NY Acad Sci 1991; 633: 615–616.
Franklin RJ . Remyelination by transplanted olfactory ensheathing cells. Anat Rec 2003; 271B: 71–76.
Franklin RJ . Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci 2002; 3: 705–714.
Kocsis JD, Akiyama Y, Lankford KL, Radtke C . Cell transplantation of peripheral-myelin-forming cells to repair the injured spinal cord. J Rehabil Res Dev 2002; 39: 287–298.
Devon R, Doucette R . Olfactory ensheathing cells myelinate dorsal root ganglion neurites. Brain Res 1992; 589: 175–179.
Ramon-Cueto A, Perez J, Nieto-Sampedro M . In vitro enfolding of olfactory neurites by p75 NGF receptor positive ensheathing cells from adult rat olfactory bulb. Eur J Neurosci 1993; 5: 1172–1180.
Plant GW et al. Purified adult ensheathing glia fail to myelinate axons under culture conditions that enable Schwann cells to form myelin. J Neurosci 2002; 22: 6083–6091.
Blakemore WF, Patterson RC . Suppression of remyelination in the CNS by X-irradiation. Acta Neuropathol (Berl) 1978; 42: 105–113.
Franklin RJ, Gilson JM, Franceschini IA, Barnett SC . Schwann cell-like myelination following transplantation of an olfactory bulb-ensheathing cell line into areas of demyelination in the adult CNS. Glia 1996; 17: 217–224.
Imaizumi T, Lankford KL, Waxman SG, Greer CA, Kocsis JD . Transplanted olfactory ensheathing cells remyelinate and enhance axonal conduction in the demyelinated dorsal columns of the rat spinal cord. J Neurosci 1998; 18: 6176–6185.
Kato T, Honmou O, Uede T, Hashi K, Kocsis JD . Transplantation of human olfactory ensheathing cells elicits remyelination of demyelinated rat spinal cord. Glia 2000; 30: 209–218.
Smith PM, Sim FJ, Barnett SC, Franklin RJ . SCIP/Oct-6, Krox-20, and desert hedgehog mRNA expression during CNS remyelination by transplanted olfactory ensheathing cells. Glia 2001; 36: 342–353.
Smith PM, Lakatos A, Barnett SC, Jeffery ND, Franklin RJ . Cryopreserved cells isolated from the adult canine olfactory bulb are capable of extensive remyelination following transplantation into the adult rat CNS. Exp Neurol 2002; 176: 402–406.
Lakatos A, Smith PM, Barnett SC, Franklin RJ . Meningeal cells enhance limited CNS remyelination by transplanted olfactory ensheathing cells. Brain 2003; 126: 598–609.
Radtke C et al. Remyelination of the nonhuman primate spinal cord by transplantation of H-transferase transgenic adult pig olfactory ensheathing cells. FASEB J 2004; 18: 335–337.
Blakemore WF, Crang AJ . The use of cultured autologous Schwann cells to remyelinate areas of persistent demyelination in the central nervous system. J Neurol Sci 1985; 70: 207–223.
Franklin RJ, Crang AJ, Blakemore WF . Type 1 astrocytes fail to inhibit Schwann cell remyelination of CNS axons in the absence of cells of the O-2A lineage. Dev Neurosci 1992; 14: 85–92.
Blesch A, Tuszynski MH . Cellular GDNF delivery promotes growth of motor and dorsal column sensory axons after partial and complete spinal cord transections and induces remyelination. J Comp Neurol 2003; 467: 403–417.
Jasmin L, Janni G, Moallem TM, Lappi DA, Ohara PT . Schwann cells are removed from the spinal cord after effecting recovery from paraplegia. J Neurosci 2000; 20: 9215–9223.
Keirstead HS, Levine JM, Blakemore WF . Response of the oligodendrocyte progenitor cell population (defined by NG2 labelling) to demyelination of the adult spinal cord. Glia 1998; 22: 161–170.
McTigue DM, Wei P, Stokes BT . Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J Neurosci 2001; 21: 3392–3400.
Watanabe M, Toyama Y, Nishiyama A . Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J Neurosci Res 2002; 69: 826–836.
McDonald JW et al. Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med 1999; 5: 1410–1412.
Sasaki M, Honmou O, Akiyama Y, Uede T, Hashi K, Kocsis JD . Transplantation of an acutely isolated bone marrow fraction repairs demyelinated adult rat spinal cord axons. Glia 2001; 35: 26–34.
Akiyama Y, Radtke C, Kocsis JD . Remyelination of the rat spinal cord by transplantation of identified bone marrow stromal cells. J Neurosci 2002; 22: 6623–6630.
Inoue M, Honmou O, Oka S, Houkin K, Hashi K, Kocsis JD . Comparative analysis of remyelinating potential of focal and intravenous administration of autologous bone marrow cells into the rat demyelinated spinal cord. Glia 2003; 44: 111–118.
Sherratt RM, Bostock H, Sears TA . Effects of 4-aminopyridine on normal and demyelinated mammalian nerve fibres. Nature 1980; 283: 570–572.
Targ EF, Kocsis JD . 4-Aminopyridine leads to restoration of conduction in demyelinated rat sciatic nerve. Brain Res 1985; 328: 358–361.
Blight AR . Effect of 4-aminopyridine on axonal conduction-block in chronic spinal cord injury. Brain Res Bull 1989; 22: 47–52.
Shi R, Blight AR . Compression injury of mammalian spinal cord in vitro and the dynamics of action potential conduction failure. J Neurophysiol 1996; 76: 1572–1580.
Shi R, Kelly TM, Blight AR . Conduction block in acute and chronic spinal cord injury: different dose–response characteristics for reversal by 4-aminopyridine. Exp Neurol 1997; 148: 495–501.
Potter PJ et al. Randomized double-blind crossover trial of fampridine-SR (sustained release 4-aminopyridine) in patients with incomplete spinal cord injury. J Neurotrauma 1998; 15: 837–849.
Jensen JM, Shi R . Effects of 4-aminopyridine on stretched mammalian spinal cord: the role of potassium channels in axonal conduction. J Neurophysiol 2003; 90: 2334–2340.
Calford MB . Dynamic representational plasticity in sensory cortex. Neuroscience 2002; 111: 709–738.
Calford MB . Mechanisms for acute changes in sensory maps. Adv Exp Med Biol 2002; 508: 451–460.
Raineteau O, Schwab ME . Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci 2001; 2: 263–273.
Basbaum AI, Wall PD . Chronic changes in the response of cells in adult cat dorsal horn following partial deafferentation: the appearance of responding cells in a previously non-responsive region. Brain Res 1976; 116: 181–204.
Darian-Smith C, Gilbert CD . Axonal sprouting accompanies functional reorganization in adult cat striate cortex. Nature 1994; 368: 737–740.
Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME . The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci 2004; 7: 269–277.
van Meeteren NL, Brakkee JH, Hamers FP, Helders PJ, Gispen WH . Exercise training improves functional recovery and motor nerve conduction velocity after sciatic nerve crush lesion in the rat. Arch Phys Med Rehabil 1997; 78: 70–77.
Bizzi E, Tresch MC, Saltiel P, d'Avella A . New perspectives on spinal motor systems. Nat Rev Neurosci 2000; 1: 101–108.
Eidelberg E, Walden JG, Nguyen LH . Locomotor control in macaque monkeys. Brain 1981; 104: 647–663.
Eidelberg E, Story JL, Walden JG, Meyer BL . Anatomical correlates of return of locomotor function after partial spinal cord lesions in cats. Exp Brain Res 1981; 42: 81–88.
Steeves JD, Tetzlaff W . Engines, accelerators, and brakes on functional spinal cord repair. Ann NY Acad Sci 1998; 860: 412–424.
Barbeau H, Rossignol S . Recovery of locomotion after chronic spinalization in the adult cat. Brain Res 1987; 412: 84–95.
Gazula VR, Roberts M, Luzzio C, Jawad AF, Kalb RG . Effects of limb exercise after spinal cord injury on motor neuron dendrite structure. J Comp Neurol 2004; 476: 130–145.
de Leon RD, Hodgson JA, Roy RR, Edgerton VR . Locomotor capacity attributable to step training versus spontaneous recovery after spinalization in adult cats. J Neurophysiol 1998; 79: 1329–1340.
de Leon RD, Hodgson JA, Roy RR, Edgerton VR . Retention of hindlimb stepping ability in adult spinal cats after the cessation of step training. J Neurophysiol 1999; 81: 85–94.
Steeves JD, Jordan LM . Localization of a descending pathway in the spinal cord which is necessary for controlled treadmill locomotion. Neurosci Lett 1980; 20: 283–288.
Loy DN et al. Functional redundancy of ventral spinal locomotor pathways. J Neurosci 2002; 22: 315–323.
Basso DM . Neuroanatomical substrates of functional recovery after experimental spinal cord injury: implications of basic science research for human spinal cord injury. Phys Ther 2000; 80: 808–817.
Wernig A, Muller S, Nanassy A, Cagol E . Laufband therapy based on ‘rules of spinal locomotion’ is effective in spinal cord injured persons. Eur J Neurosci 1995; 7: 823–829.
Wernig A, Nanassy A, Muller S . Maintenance of locomotor abilities following Laufband (treadmill) therapy in para- and tetraplegic persons: follow-up studies. Spinal Cord 1998; 36: 744–749.
Wirz M, Colombo G, Dietz V . Long term effects of locomotor training in spinal humans. J Neurol Neurosurg Psychiatr 2001; 71: 93–96.
Bavetta S, Hamlyn PJ, Burnstock G, Lieberman AR, Anderson PN . The effects of FK506 on dorsal column axons following spinal cord injury in adult rats: neuroprotection and local regeneration. Exp Neurol 1999; 158: 382–393.
Constantini S, Young W . The effects of methylprednisolone and the ganglioside GM1 on acute spinal cord injury in rats. J Neurosurg 1994; 80: 97–111.
De Ley G, Leybaert L . Effect of flunarizine and methylprednisolone on functional recovery after experimental spinal injury. J Neurotrauma 1993; 10: 25–35.
Diaz-Ruiz A et al. Lipid peroxidation inhibition in spinal cord injury: cyclosporin-A vs methylprednisolone. Neuroreport 2000; 11: 1765–1767.
Haghighi SS et al. Effects of methylprednisolone and MK-801 on functional recovery after experimental chronic spinal cord injury. Spinal Cord 2000; 38: 733–740.
Kim DH, Jahng TA . Continuous brain-derived neurotrophic factor (BDNF) infusion after methylprednisolone treatment in severe spinal cord injury. J Korean Med Sci 2004; 19: 113–122.
Lankhorst AJ, ter Laak MP, Hamers FP, Gispen WH . Combined treatment with alphaMSH and methylprednisolone fails to improve functional recovery after spinal injury in the rat. Brain Res 2000; 859: 334–340.
Mu X, Azbill RD, Springer JE . Riluzole and methylprednisolone combined treatment improves functional recovery in traumatic spinal cord injury. J Neurotrauma 2000; 17: 773–780.
Sheng H, Spasojevic I, Warner DS, Batinic-Haberle I . Mouse spinal cord compression injury is ameliorated by intrathecal cationic manganese(III) porphyrin catalytic antioxidant therapy. Neurosci Lett 2004; 366: 220–225.
Takami T, Oudega M, Bethea JR, Wood PM, Kleitman N, Bunge MB . Methylprednisolone and interleukin-10 reduce gray matter damage in the contused Fischer rat thoracic spinal cord but do not improve functional outcome. J Neurotrauma 2002; 19: 653–666.
Ibarra A, Hauben E, Butovsky O, Schwartz M . The therapeutic window after spinal cord injury can accommodate T cell-based vaccination and methylprednisolone in rats. Eur J Neurosci 2004; 19: 2984–2990.
Baffour R et al. Synergistic effect of basic fibroblast growth factor and methylprednisolone on neurological function after experimental spinal cord injury. J Neurosurg 1995; 83: 105–110.
Li L, Xu Q, Wu Y, Hu W, Gu P, Fu Z . Combined therapy of methylprednisolone and brain-derived neurotrophic factor promotes axonal regeneration and functional recovery after spinal cord injury in rats. Chin Med J (Engl) 2003; 116: 414–418.
Haghighi SS, Clapper A, Johnson GC, Stevens A, Prapaisilp A . Effect of 4-aminopyridine and single-dose methylprednisolone on functional recovery after a chronic spinal cord injury. Spinal Cord 1998; 36: 6–12.
Guth L, Zhang Z, Roberts E . Key role for pregnenolone in combination therapy that promotes recovery after spinal cord injury. Proc Natl Acad Sci USA 1994; 91: 12308–12312.
Kuchner EF, Hansebout RR . Combined steroid and hypothermia treatment of experimental spinal cord injury. Surg Neurol 1976; 6: 371–376.
Kuchner EF, Hansebout RR, Pappius HM . Effects of dexamethasone and of local hypothermia on early and late tissue electrolyte changes in experimental spinal cord injury. J Spinal Disord 2000; 13: 391–398.
Yamamoto K, Ishikawa T, Sakabe T, Taguchi T, Kawai S, Marsala M . The hydroxyl radical scavenger Nicaraven inhibits glutamate release after spinal injury in rats. Neuroreport 1998; 9: 1655–1659.
Yick LW, So KF, Cheung PT, Wu WT . Lithium chloride reinforces the regeneration-promoting effect of chondroitinase ABC on rubrospinal neurons after spinal cord injury. J Neurotrauma 2004; 21: 932–943.
Wang MS, Gold BG . FK506 increases the regeneration of spinal cord axons in a predegenerated peripheral nerve autograft. J Spinal Cord Med 1999; 22: 287–296.
von Meyenburg J, Brosamle C, Metz GA, Schwab ME . Regeneration and sprouting of chronically injured corticospinal tract fibers in adult rats promoted by NT-3 and the mAb IN-1, which neutralizes myelin-associated neurite growth inhibitors. Exp Neurol 1998; 154: 583–594.
Azanchi R, Bernal G, Gupta R, Keirstead HS . Combined demyelination plus Schwann cell transplantation therapy increases spread of cells and axonal regeneration following contusion injury. J Neurotrauma 2004; 21: 775–788.
Keirstead HS, Morgan SV, Wilby MJ, Fawcett JW . Enhanced axonal regeneration following combined demyelination plus schwann cell transplantation therapy in the injured adult spinal cord. Exp Neurol 1999; 159: 225–236.
Yick LW, Wu W, So KF, Yip HK, Shum DK . Chondroitinase ABC promotes axonal regeneration of Clarke's neurons after spinal cord injury. Neuroreport 2000; 11: 1063–1067.
King VR, Phillips JB, Brown RA, Priestley JV . The effects of treatment with antibodies to transforming growth factor beta1 and beta2 following spinal cord damage in the adult rat. Neuroscience 2004; 126: 173–183.
Oudega M, Hagg T . Nerve growth factor promotes regeneration of sensory axons into adult rat spinal cord. Exp Neurol 1996; 140: 218–229.
Richardson PM, Verge VM . The induction of a regenerative propensity in sensory neurons following peripheral axonal injury. J Neurocytol 1986; 15: 585–594.
Chong MS, Woolf CJ, Turmaine M, Emson PC, Anderson PN . Intrinsic versus extrinsic factors in determining the regeneration of the central processes of rat dorsal root ganglion neurons: the influence of a peripheral nerve graft. J Comp Neurol 1996; 370: 97–104.
Oudega M, Varon S, Hagg T . Regeneration of adult rat sensory axons into intraspinal nerve grafts: promoting effects of conditioning lesion and graft predegeneration. Exp Neurol 1994; 129: 194–206.
Blits B, Dijkhuizen PA, Boer GJ, Verhaagen J . Intercostal nerve implants transduced with an adenoviral vector encoding neurotrophin-3 promote regrowth of injured rat corticospinal tract fibers and improve hindlimb function. Exp Neurol 2000; 164: 25–37.
Fernandez E, Pallini R, Mercanti D . Effects of topically administered nerve growth factor on axonal regeneration in peripheral nerve autografts implanted in the spinal cord of rats. Neurosurgery 1990; 26: 37–42.
Hiebert GW, Khodarahmi K, McGraw J, Steeves JD, Tetzlaff W . Brain-derived neurotrophic factor applied to the motor cortex promotes sprouting of corticospinal fibers but not regeneration into a peripheral nerve transplant. J Neurosci Res 2002; 69: 160–168.
Kwon BK, Liu J, Oschipok L, Teh J, Liu ZW, Tetzlaff W . Rubrospinal neurons fail to respond to brain-derived neurotrophic factor applied to the spinal cord injury site 2 months after cervical axotomy. Exp Neurol 2004; 189: 45–57.
Levi AD, Dancausse H, Li X, Duncan S, Horkey L, Oliviera M . Peripheral nerve grafts promoting central nervous system regeneration after spinal cord injury in the primate. J Neurosurg Spine 2002; 96: 197–205.
Novikova LN, Novikov LN, Kellerth JO . Differential effects of neurotrophins on neuronal survival and axonal regeneration after spinal cord injury in adult rats. J Comp Neurol 2002; 452: 255–263.
Storer PD, Houle JD, Oblinger M, Jones KJ . Combination of gonadal steroid treatment and peripheral nerve grafting results in a peripheral motoneuron-like pattern of beta II-tubulin mRNA expression in axotomized hamster rubrospinal motoneurons. J Comp Neurol 2002; 449: 364–373.
Ye JH, Houle JD . Treatment of the chronically injured spinal cord with neurotrophic factors can promote axonal regeneration from supraspinal neurons. Exp Neurol 1997; 143: 70–81.
Bamber NI, Li H, Lu X, Oudega M, Aebischer P, Xu XM . Neurotrophins BDNF and NT-3 promote axonal re-entry into the distal host spinal cord through Schwann cell-seeded mini-channels. Eur J Neurosci 2001; 13: 257–268.
Blits B, Oudega M, Boer GJ, Bartlett BM, Verhaagen J . Adeno-associated viral vector-mediated neurotrophin gene transfer in the injured adult rat spinal cord improves hind-limb function. Neuroscience 2003; 118: 271–281.
Iannotti C, Li H, Yan P, Lu X, Wirthlin L, Xu XM . Glial cell line-derived neurotrophic factor-enriched bridging transplants promote propriospinal axonal regeneration and enhance myelination after spinal cord injury. Exp Neurol 2003; 183: 379–393.
Oudega M, Xu XM, Guenard V, Kleitman N, Bunge MB . A combination of insulin-like growth factor-I and platelet-derived growth factor enhances myelination but diminishes axonal regeneration into Schwann cell grafts in the adult rat spinal cord. Glia 1997; 19: 247–258.
Tuszynski MH, Weidner N, McCormack M, Miller I, Powell H, Conner J . Grafts of genetically modified Schwann cells to the spinal cord: survival, axon growth, and myelination. Cell Transplant 1998; 7: 187–196.
Weidner N, Blesch A, Grill RJ, Tuszynski MH . Nerve growth factor-hypersecreting Schwann cell grafts augment and guide spinal cord axonal growth and remyelinate central nervous system axons in a phenotypically appropriate manner that correlates with expression of L1. J Comp Neurol 1999; 413: 495–506.
Bakshi A, Fisher O, Dagci T, Himes BT, Fischer I, Lowman A . Mechanically engineered hydrogel scaffolds for axonal growth and angiogenesis after transplantation in spinal cord injury. J Neurosurg Spine 2004; 1: 322–329.
Bloch J, Fine EG, Bouche N, Zurn AD, Aebischer P . Nerve growth factor- and neurotrophin-3-releasing guidance channels promote regeneration of the transected rat dorsal root. Exp Neurol 2001; 172: 425–432.
Houle JD . Regeneration of dorsal root axons is related to specific non-neuronal cells lining NGF-treated intraspinal nitrocellulose implants. Exp Neurol 1992; 118: 133–142.
Houle JD, Ziegler MK . Bridging a complete transection lesion of adult rat spinal cord with growth factor-treated nitrocellulose implants. J Neural Transplant Plast 1994; 5: 115–124.
Prewitt CM, Niesman IR, Kane CJ, Houle JD . Activated macrophage/microglial cells can promote the regeneration of sensory axons into the injured spinal cord. Exp Neurol 1997; 148: 433–443.
Tobias CA, Dhoot NO, Wheatley MA, Tessler A, Murray M, Fischer I . Grafting of encapsulated BDNF-producing fibroblasts into the injured spinal cord without immune suppression in adult rats. J Neurotrauma 2001; 18: 287–301.
Bregman BS, Broude E, McAtee M, Kelley MS . Transplants and neurotrophic factors prevent atrophy of mature CNS neurons after spinal cord injury. Exp Neurol 1998; 149: 13–27.
Broude E, McAtee M, Kelley MS, Bregman BS . Fetal spinal cord transplants and exogenous neurotrophic support enhance c-Jun expression in mature axotomized neurons after spinal cord injury. Exp Neurol 1999; 155: 65–78.
Castellanos DA, Tsoulfas P, Frydel BR, Gajavelli S, Bes JC, Sagen J . TrkC overexpression enhances survival and migration of neural stem cell transplants in the rat spinal cord. Cell Transplant 2002; 11: 297–307.
Han PJ, Shukla S, Subramanian PS, Hoffman PN . Cyclic AMP elevates tubulin expression without increasing intrinsic axon growth capacity. Exp Neurol 2004; 189: 293–302.
Gautier SE et al. Poly(alpha-hydroxyacids) for application in the spinal cord: resorbability and biocompatibility with adult rat Schwann cells and spinal cord. J Biomed Mater Res 1998; 42: 642–654.
Guenard V, Kleitman N, Morrissey TK, Bunge RP, Aebischer P . Syngeneic Schwann cells derived from adult nerves seeded in semipermeable guidance channels enhance peripheral nerve regeneration. J Neurosci 1992; 12: 3310–3320.
Novikov LN, Novikova LN, Mosahebi A, Wiberg M, Terenghi G, Kellerth JO . A novel biodegradable implant for neuronal rescue and regeneration after spinal cord injury. Biomaterials 2002; 23: 3369–3376.
Oudega M et al. Axonal regeneration into Schwann cell grafts within resorbable poly(alpha-hydroxyacid) guidance channels in the adult rat spinal cord. Biomaterials 2001; 22: 1125–1136.
Paino CL, Bunge MB . Induction of axon growth into Schwann cell implants grafted into lesioned adult rat spinal cord. Exp Neurol 1991; 114: 254–257.
Paino CL, Fernandez-Valle C, Bates ML, Bunge MB . Regrowth of axons in lesioned adult rat spinal cord: promotion by implants of cultured Schwann cells. J Neurocytol 1994; 23: 433–452.
Schlosshauer B, Muller E, Schroder B, Planck H, Muller HW . Rat Schwann cells in bioresorbable nerve guides to promote and accelerate axonal regeneration. Brain Res 2003; 963: 321–326.
Chauhan NB, Figlewicz HM, Khan T . Carbon filaments direct the growth of postlesional plastic axons after spinal cord injury. Int J Dev Neurosci 1999; 17: 255–264.
Dupont-Versteegden EE, Murphy RJ, Houle JD, Gurley CM, Peterson CA . Mechanisms leading to restoration of muscle size with exercise and transplantation after spinal cord injury. Am J Physiol Cell Physiol 2000; 279: C1677–C1684.
Peterson CA, Murphy RJ, Dupont-Versteegden EE, Houle JD . Cycling exercise and fetal spinal cord transplantation act synergistically on atrophied muscle following chronic spinal cord injury in rats. Neurorehabil Neural Repair 2000; 14: 85–91.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Ramer, L., Ramer, M. & Steeves, J. Setting the stage for functional repair of spinal cord injuries: a cast of thousands. Spinal Cord 43, 134–161 (2005). https://doi.org/10.1038/sj.sc.3101715
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.sc.3101715
Keywords
This article is cited by
-
Transplantation of Cerebral Dopamine Neurotrophic Factor Transducted BMSCs in Contusion Spinal Cord Injury of Rats: Promotion of Nerve Regeneration by Alleviating Neuroinflammation
Molecular Neurobiology (2016)
-
Nanoparticle-Hydrogel: A Hybrid Biomaterial System for Localized Drug Delivery
Annals of Biomedical Engineering (2016)
-
The efficacy of neuromuscular electrical stimulation with alternating currents in the kilohertz frequency to stimulate gait rhythm in rats following spinal cord injury
BioMedical Engineering OnLine (2015)
-
Ethics, health care and spinal cord injury: research, practice and finance
Spinal Cord (2011)
-
Neurotrophins: Potential Therapeutic Tools for the Treatment of Spinal Cord Injury
Neurotherapeutics (2011)
