
Abstract
We assessed the effects of i.v. cocaine on parasympathetic and sympathetic nervous system activity, and on the complexity vs. regularity of changes in heart rate over time. Fourteen otherwise healthy men with histories of i.v. cocaine abuse received bolus injections of cocaine (20 mg or 40 mg) and placebo (saline) on different days. Cardiovascular measures derived from the electrocardiogram, including heart rate, Porges’ vagal tone (respiratory sinus arrhythmia), the 0.10 Hz rhythm, Toichi's vagal index, Toichi's sympathetic index, and approximate entropy (ApEn), were measured continuously. As predicted, cocaine produced tachycardia, accompanied by pronounced decreases in response to 40 mg cocaine in two different vagal tone indexes that precisely mirrored the increases in heart rate. The measure of sympathetic (and vagal) neural influences on the heart (0.10 Hz wave) also decreased in response to cocaine. Converging evidence from Toichi's vagal index supported the conclusion that the tachycardia from cocaine was due to withdrawal of cardiac vagal tone. These findings, and evidence that cocaine decreased cardiovascular complexity, contradict the prevailing assumption that the mechanism by which cocaine produces tachycardia is sympathetic (beta-adrenergic). We discuss implications for cardiac arrhythmias associated with cocaine abuse and death due to overdose.
Similar content being viewed by others


Main
Cocaine is a widely abused drug with pronounced subjective and cardiovascular effects (Fischman et al. 1976; Johanson and Fischman 1989). Early human research on cocaine (Fischman et al. 1976; Resnick et al. 1977) revealed dose-dependent positive chronotropic effects on the heart, reaching a mean increase in heart rate of 38 bpm in response to 32 mg i.v. cocaine. Effects on the cardiovascular system can be remarkable in some subjects. Increases in heart rate have been reported to occur in response to doses as low as 13 mg i.v. cocaine; these increases ranged from 15 to 46 bpm (Ambre et al. 1988). The magnitude of these effects has led to the conclusion that cardiotoxicity from cocaine may be a major factor in death due to cocaine overdose (Billman 1990).
The cardiotoxicity of cocaine has directed interest toward the autonomic mechanism by which the drug produces cardiovascular arousal. It has been assumed (Billman 1990; Ritchie and Greene 1990) that the cardiovascular response to cocaine is due primarily to activation of sympathetic autonomic mechanisms. This view stems in part from evidence that cocaine directly blocks reuptake of catecholamines in the periphery (Ritchie and Greene 1990). Clinical case reports have suggested that propranolol (a beta-adrenergic blocker) and phentolamine (an alpha-adrenergic blocker) may partially reverse cardiovascular arousal in cocaine overdose (Rappolt et al. 1977). However, controlled studies at moderate dosages of cocaine in humans would be needed to confirm these observations. Furthermore, there is conflicting evidence in animal research concerning the role of the sympathetic nervous system in the response to cocaine in the intact organism. Recent findings indicate that central administration of cocaine actually decreases peripheral sympathetic neural activity in cats and dogs (Raczkowski et al. 1991).
Tachycardia can occur for different reasons. An increase in sympathetic drive would increase heart rate (Berne and Levy 1977). This effect has been the presumed mechanism of the tachycardic response to cocaine (Ritchie and Greene 1990). However, tachycardia can also result from withdrawal of tonic vagal inhibition (Berne and Levy 1977). Vagal blockade in humans almost doubles resting heart rate (Dellinger et al. 1987), indicating that there is profound tonic suppression of heart rate by cardiac vagal tone via the parasympathetic nervous system. This effect would preclude using vagal blockade to determine whether the tachycardia produced by cocaine could be blocked by a drug such as atropine because massive baseline shifts in resting heart rate would obscure interpretation of the results. Also, i.v. cocaine could not be administered to human subjects with very high resting heart rates (following atropine). Recent findings that cocaine can block muscarinic M2 receptors in the heart and brain (Sharkey et al. 1988) implicates the parasympathetic nervous system, including effects on vagal tone, in the cardiotoxicity of cocaine.
The recent development of vagal tone as a noninvasive measure of vagal inhibition of the heart (Porges, 1995) allowed us to measure parasympathetic influences in response to intravenous cocaine in humans. Vagal tone is derived from the electrocardiogram (ECG) using time series analysis of successive R-wave to R-wave intervals. It quantifies respiratory sinus arrhythmia, or normal heart rate variability in the respiratory frequency band. This variability has been viewed as a relatively pure measure of cardiac vagal tone (Porges and Bohrer 1990). Moreover, there is evidence (Axelrod et al. 1981) that the 0.10 Hz rhythm, measured analogously to vagal tone but at a lower frequency, reflects mixed influences, including (but not limited to) beta-adrenergic effects on the heart.
In addition, recently developed chaos and complexity measures were used in the present study to: (1) provide converging evidence concerning autonomic mechanisms of cocaine's cardiovascular effects; (2) measure aspects of system function that were relevant to the cardiotoxicity of cocaine; and (3) assess the effect of cocaine on cardiovascular system dynamics. Toichi et al. (1997) derived measures of vagal and sympathetic function based on the Lorenz plots of successive R-wave to R-wave intervals in the ECG. The Lorenz plot has each R–R interval on the Y-axis and the next successive R–R interval on the X-axis. The vagal and sympathetic indexes were selected empirically by Toichi et al. (1997) as those functions from the Lorenz plots that were most sensitive to vagal and sympathetic blockade with atropine and propranolol, respectively.
Pincus et al.'s (1991) Approximate Entropy (ApEn) is a measure of the regularity versus complexity of a time series. As applied to successive R-wave to R-wave intervals, ApEn quantifies the degree to which the cardiac series can be characterized by simple repeating patterns (regularity) versus many different patterns that repeat less frequently (complexity). Pincus and Goldberger (1994) argued that disease states are associated with highly regular series (i.e., low ApEn) while “healthy” biological systems tend to be complex (i.e., high ApEn). The mathematical approach to ApEn is very different from that of Porges’ Vagal Tone or Toichi's Vagal and Sympathetic Indexes, although they all concern variability as opposed to mean level of heart rate.
We assessed a number of cardiac measures that are derived from the ECG in response to i.v. administration of cocaine or placebo in order to determine the autonomic mechanisms of the cardiovascular response to cocaine.
METHODS
Subjects
Fourteen healthy male subjects completed the protocol (one subject failed to complete because of an ECG abnormality that prevented him from receiving cocaine). These are the same subjects reported in brief form in Newlin (1995), and the subjects partially overlap those of a positron emission tomography study (London et al. 1990) of the effects of cocaine on brain function. The subjects ranged in age from 25 to 43 years (mean age = 31.6). Eight of the subjects were African-American, and six were whites of European descent. None of these men were seeking or receiving treatment for cocaine abuse.
Subjects had no access to cocaine (other than the i.v. injections reported here) or to other non-prescribed drugs (other than nicotine and caffeine) for at least one week while residing on a residential research ward prior to initiating the protocol, and they resided there for the duration of the study. They received a physical examination prior to the study, with particular attention paid to ruling out subjects with cardiac abnormalities. A resting 12-lead electrocardiograph and 3 min rhythm strips were used to exclude subjects with any significant past injury or present ischemia, or any other condition that would preclude their receiving cocaine. Resting blood pressure had to be within normal limits (<140 mmHg systolic and <90 diastolic).
Most of the subjects were polydrug abusers. All subjects had reported histories of i.v. cocaine abuse and/or dependence, and had histories of urinalysis results that were positive for cocaine or its metabolites. Twelve subjects reported having used i.v. cocaine within the last 14 days, and seven had used heroin in that period. Subjects reported average cocaine use of 0.53 g per day (two subjects reported using $90.00 per week of cocaine and $45.00 per month, respectively). They had used cocaine for a mean duration of 9 years (range: 2–20 years). Eleven of the subjects were current cigarette smokers, and nine subjects had an alcohol abuse or dependence diagnosis.
All subjects signed a consent form approved by the Francis Scott Key Hospital Institutional Review Board of Johns Hopkins Medical Institutions. This consent form informed them of the risks of receiving i.v. cocaine or placebo. Subjects were paid volunteers.
Cocaine
Cocaine HCl or placebo (0.9% NaCl) was administered intravenously on three separate days at least 24 h apart with one of the following doses per day: 0 mg cocaine (placebo), 20 mg cocaine, and 40 mg cocaine. Bolus injections were 10 s in duration. The experimental team and the subjects were not aware of the order of the dosages. The dosages were in pseudo-randomized order, with the 20-mg dose of cocaine always preceding the 40-mg dose to enhance safety. The placebo dose could occur in any place in the sequence.
Procedure
Cocaine and placebo were administered by a physician in a sound-attenuated chamber. The subjects were blind-folded, and they were seated, listening to an audiotape over headphones that presented white noise, a series of questions given intermittently, and “beep” prompts (London et al. 1990). Three experimental sessions were preceded by a single-blind administration of placebo to familiarize the subjects with the environment and to overcome first-session effects (Newlin and Pretorius 1991).
A signal that the injection was imminent was the entry of a physician into the chamber. After a mean of 64 s, the physician injected the solution into the port of an indwelling venous catheter in the subject's arm. A technician was seated in the subject chamber during the entire recording period, both before and after the injection. However, the physician's entry into the room was still highly predictive of the imminent injection.
Dependent Measures
Heart rate, vagal tone index (Porges 1995), and the 0.10 Hz rhythm were recorded continuously for 5 min before the physician entered the room, during the time the physician was in the room but before the injection, and for 30 min after the injection (London et al. 1990).
Vagal tone and the 0.10 Hz rhythm were quantified using patented (Porges 1995) software (MXEDIT) from Delta-Biometrics (Bethesda, MD). This software takes as input the successive R-wave to R-wave intervals in ms. A technician, who was blinded to the treatment (placebo or 20 mg or 40 mg cocaine), performed computerized editing on this train of intervals in order to remove artifact and to recover the original heart rate signal. Mxedit quantifies vagal tone in adult subjects from a heart period time series, includes the following steps: (1) heart period data is resampled every 500 ms; (2) a 21-point moving cubic polynomial filter is stepped through the time sampled series to produce a smoothed template series; (3) the template series is subtracted from the original series to produce a residual time series; (4) a digital bandpass filter with 25 coefficients is stepped through the residual series to extract variance in the .12–.40 Hz frequency band; and, (5) the remaining variance is transformed by its natural logarithm, and is used as a measure of vagal tone. The smoothing techniques contained in these steps function to remove variance in the heart period data with frequency characteristics that fall outside the frequency band of spontaneous respiration, allowing for accurate quantification of the amplitude of respiratory sinus arrhythmia, or vagal tone. Similar quantification procedures were used for the 0.10 Hz rhythm, but the frequency band of interest was 0.06 to 0.10 Hz. The n was 14 for these analyses. The 0.10 Hz rhythm reflects activity at the baroreceoptor reflex feedback frequency rather than respiratory sinus arrhythmia (for vagal tone).
Chaos-related measures of the Vagal Index (sensitive to pharmacological blockade with atropine) and the Sympathetic Index (sensitive to blockade by propranolol) were derived from the train of successive R-wave to R-wave intervals from the ECG, using the method described by Toichi and colleagues (1997). Briefly, the Lorenz plot was constructed in which each adjacent R-wave to R-wave interval was plotted against the next R-wave to R-wave interval. This transformation yielded a scatterplot such as that shown in Figure 1 . The plot was rotated using trigonometric functions so that the diagonal axis became the X-axis. The standard deviation of differences along the X- and Y-axes of the rotated Lorenz plot were then calculated, yielding Toichi's L and T, respectively. The Vagal Index was calculated as the log of (L × T), and the Sympathetic Index was taken as L divided by T. The n was equal to 9 for these analyses because some subjects’ raw data were deemed too artifact-ridden to perform the transformations described above.

Lorenz plots of each successive R–R interval (minus the mean R–R interval for the series) against the next interval, which were used to calculate Toichi's Vagal Index (CVI) and his Sympathetic Index (CSI). The diagonal lines represent axes which were rotated to calculate Toichi's indexes (see text). The upper trace (A) is prior to cocaine, and the lower trace (B) is 8 min after 40 mg i.v. cocaine in one subject. Note that cocaine markedly reduced scatter in the Lorenz plot of this particular subject
The dependent measures in these analyses represented the means for each subject for study intervals 3 to 24 min after injection of cocaine minus the 5 min pre-drug baseline mean. Therefore, it was the mean post-injection minus the pre-drug baseline level. Note that this transformation was different from that concerning vagal tone and the 0.10 Hz rhythm.
Approximate Entropy (ApEn) was also calculated on the successive R-wave to R-wave intervals for each study interval using the method described by Pincus et al. (1991). The algorithm was tested for accuracy using pieces of data upon which ApEn had been calculated using Pincus’ own software. The parameter “m” concerns the length of “runs” (i.e., number of R–R intervals) on which the next value (i.e., R–R interval) is compared in Pincus’ algorithm, and the parameter “r” is a “filter” for the magnitude of change (in R–R interval) that is needed to identity a change in pattern of successive intervals. Results for m = 1 and m = 2 are presented separately. The statistic r was based for each subject on 0.2 times the standard deviation of R–R intervals during their 5 min baseline prior to placebo. This standardized the window used in the calculation of ApEn for each subject (r did not change between sessions) although it meant that each subject had a different value of r.
Part of the data on one subject from the double-blind placebo session was lost due to equipment malfunction. Several data points, therefore were interpolated during the stable, final portion of the post-injection period. The n was 9 for these analyses.
Statistical Methods
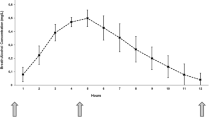
Mean heart rate, vagal tone index, and the 0.10 Hz rhythm were quantified for the interval prior to the physician entering the room, the period in which the physician was in the room but before the injection, and for 15 successive 2 min intervals after injection (Figure 2). Statistical significance of the effect of cocaine-related cues was tested with repeated-measures analysis of variance (ANOVA) on the difference between the 5 min baseline and the mean of the period in which the physician was in the experimental chamber with the subject. The effect of placebo was tested with repeated-measures ANOVA on the difference between the 5 min baseline and the first 2 min block after the injection of placebo.

Responses in heart rate, Porges’ vagal tone, and the 0.10 Hz rhythm to intravenous cocaine (20 mg and 40 mg) or placebo in 14 cocaine abusing volunteers. Shown are means (±S.E.M.) at baseline (BL) before injection, after the physician entered the subject chamber signaling imminent injection (Cues), and for 30 min after injection. The vertical dashed line indicates the point of i.v. injection. Note that cocaine (20 mg and 40 mg) significantly increased heart rate (p < .0001) and that 40 mg cocaine decreased vagal tone index (p < .01) in a dose-dependent manner. This indicated that the tachycardia from cocaine was, at least in part, due to withdrawal of vagal (parasympathetic) inhibition of the heart. Peak cardiovascular effects of cocaine occurred at approximately 7 to 11 min after injection. Effects of placebo injections were short-lived, and were most apparent in the first 2-min period after injection
Statistical significance of the effect of drug was tested using repeated-measures ANOVA. There were no significant baseline differences in heart rate (F(2,39) = 0.1, n.s.), vagal tone index (F(2,39) = 0.03, n.s.), or the 0.10 Hz rhythm (F(2,39) = 0.3, n.s.). Therefore, difference scores were constructed for each dose by subtracting the pre-injection baseline from the mean of the scores after injection. Note that the difference scores were based on the pre-drug baseline corresponding to that day's dose, so that the baselines were different for each drug condition.
The p < .01 criterion for statistical significance was adopted to correct for multiple comparisons.
RESULTS
Mean heart rate, vagal tone index, and 0.10 Hz rhythm results are displayed in Figure 2. The increase in heart rate of 6 bpm from baseline to the interval in which the physician was in the subject chamber (cues) was significant (F(1,13) = 26.3, p < .0002), but the change in vagal tone index (F(1,13) = 0.2, n.s.) and the 0.10 Hz rhythm (F(1,13) = 2.3, n.s.) were not. Similarly, the increase in heart rate of 11 bpm in the first 2 min after placebo was significant (F(1,13) = 13.8, p < .003), but the change in vagal tone index (F(1,13) = 0.5, n.s.) and in the 0.10 Hz rhythm (F(1,13) = 0.9, n.s.) were not. Heart rate returned rapidly to baseline levels after this increase in the placebo condition (Figure 2).
After the injection of cocaine, there was a robust, dose-dependent tachycardia. The increase in heart rate showed a peak of approximately 30 bpm from 7 to 11 min after the injection of 40 mg cocaine. For heart rate, the main effect of dose (40 mg, 20 mg, and placebo) was significant (F(2,39) = 37.7, p < .0001), and the contrasts between placebo and 20 mg (F(1,26) = 41.8, p < .0001), and between placebo and 40 mg (F(1,26) = 73.7, p < .0001) were significant, although that between 20 mg and 40 mg (F(1,26) = 4.7, p < .05) was not. This degree of tachycardia was consistent with previous results (Fischman et al. 1976; Resnick et al. 1977).
The increase in heart rate was mirrored temporally by an almost significant (F(2,39) = 4.6, p < .02), dose-dependent decrease in vagal tone index. The change in vagal tone also showed a trough at approximately 7 to 11 min after injection when 40 mg was administered, and it was a decrease of approximately 2 log units. Contrasts between placebo and 20 mg (F(1,26) = 8.2, p < .01), and between placebo and 40 mg cocaine (F(1,26) = 7.6), p < .01) were significant. The contrast between 20 mg and 40 mg (F(1,26) = 0.7, n.s.) was not significant. None of the measures returned entirely to baseline levels during the 30-min recording period after 40 mg cocaine.
The negative correlations between peak changes in heart rate and vagal tone index were r = −.79 under placebo, r = −0.46 at the 20-mg dose, and r = −0.74 at the 40-mg dose.
For the 0.10 Hz wave, the overall decrease following cocaine was significant (F(2,39) = 5.4, p < .01). The contrast between the placebo and 40 mg dose was significant (F(1,26) = 9.3, p < .01), but those between the placebo and 20 mg dose (F(1,26) = 2.7, n.s.) and between the 20 mg and 40 mg dosages (F(1,26) = 3.1, p < .10) were not significant.
Results concerning Toichi's Vagal Index and Sympathetic Index, and Pincus’ ApEn, with R–R interval (heart period) as a basis for comparison, are presented in Figure 3. Intravenous cocaine decreased the Vagal Index and ApEn (m = 1) and ApEn (m = 2). For Toichi's Vagal Index, the decrease from cocaine was significant (F(2,16) = 6.2, p < .01), but the small increase in the Sympathetic Index was not (F(2,16) = 2.7, p < .10). As might be expected, the changes in these two indexes (Vagal and Sympathetic Indexes) from the high dose of cocaine (minus the placebo change) were negatively correlated (r = −.46). To determine whether the cocaine-induced change in the Vagal Index could account completely for the change in heart period, we entered change in the Vagal Index (40 mg minus placebo) as a covariate in the ANCOVA for heart period. The main effect of cocaine on heart period remained significant (F(2,14) = 20.2, p < .001) after controlling for the effect of cocaine on the Vagal Index.

Mean responses (+S.E.M.) to cocaine or placebo in (A) heart period (the reciprocal of heart rate), (B) Toichi's Vagal Index (CVI) and (C) Sympathetic Index (CSI), and (D) ApEn with m = 1 or m = 2. (E) the m parameter is the length of patterns used to quantify regularity of the time series vs. complexity of the series. Each bar represents activity from 3 to 24 min after injection of cocaine (20 mg or 40 mg) or placebo, after subtracting the baseline (pre-drug) level. Note that cocaine decreased CVI (B) but had no significant effect on CSI (C). Cocaine decreased ApEn (m = 1) (D), and marginally (p < .02) decreased ApEn (m = 2) (E); this indicated that cocaine increased the regularity and decreased the complexity of the cardiovascular time series
The effect of cocaine to decrease ApEn (i.e., to reduce complexity and increase regularity) was significant for ApEn (m = 1) (F(2,16) = 14.4, p < .001) and was almost significant (F(2,16) = 5.3, p = .017) for ApEn (m = 2). Change in the two ApEn measures were substantially correlated with each other (r = .72), and change in ApEn (m = 1) was highly correlated with change in Toichi's Vagal Index (r = .90). The effect of cocaine on heart period remained significant after entering either ApEn (m = 1) (F(2,14) = 8.5, p = .004) or ApEn (m = 2) (F(2,14) = 14.2, p < .001) as covariates.
DISCUSSION
The fact that cocaine-induced changes in heart rate and vagal tone mirrored each other temporally indicated that there was a substantial vagal component to the tachycardia. Withdrawal of vagal inhibition (associated with a decrease in vagal tone) would drive heart rate higher. Studies with atropine in human subjects (Dellinger et al. 1987) have indicated that a change in vagal tone index of this magnitude is roughly equivalent to a dosage of atropine, a vagal blocker, of approximately 1 mg i.m. Decreases in vagal tone index of similar magnitude 5 min after smoking marijuana cigarettes have been noted previously (Newlin et al. 1991). Although the cardiovascular effects of morphine, alcohol, nicotine, methylphenidate, and pentobarbitol were not as pronounced, all of these substances have been reported (Newlin et al. 1990; Pretorius et al. 1991; Wong et al. 1991) to decrease vagal tone index in humans. Therefore, it is possible that withdrawal of vagal tone is a common factor in the response to abused drugs.
The effects of i.v. cocaine on heart rate and the time course (i.e., tachycardia peaking approximately 10 min after injection) are congruent with earlier studies (Fischman et al. 1976; Resnick et al. 1977). The higher dose of cocaine (40 mg) in the current study produced significant subjective effects in the positron emission tomography portion of this study (London et al. 1990) with which there was some overlap among subjects.
There is evidence from studies in squirrel monkeys that the tachycardia from i.v. cocaine can be partially blocked by either propranolol or phentolamine (Tella et al. 1990). These results would indicate that there are, in fact, sympathetic components to the increase in heart rate from cocaine, at least in monkeys. It is possible that there are important species differences in the cardiovascular response to cocaine. Hexamethonium (an autonomic ganglionic blocker) appears to block the tachycardia from cocaine in dogs (Wilkerson 1989) but not in squirrel monkeys (Tella et al. 1990).
The present results indicated that cocaine may have actually reduced sympathetic neural influences on the heart. This finding is consistent with the animal data (Gantenberg and Hageman 1991), but stands in marked contrast to prevailing assumptions concerning the effect of cocaine on beta-adrenergic influences on the heart. However, there was a slight, nonsignificant increase in Toichi's sympathetic index in response to cocaine.
The results indicated that cocaine-related cues produced an initial tachycardic response that was not vagal in origin. The responses to cocaine-related cues (i.e., the physician and the placebo injection) are important for two reasons. First, they reflect both an anticipatory and immediately post-injection placebo response that is independent of parasympathetic mechanisms. Conditioned responses to cocaine-related cues were expected in this sample of cocaine abusers due to their extensive past histories of Pavlovian conditioning to these cues. As entry of the physician was a highly predictive cue that the injection was imminent (Cascella et al. 1989; Muntaner et al. 1989), this increase in heart rate likely represented an anticipatory response to cocaine cues. Cardiovascular responses to cocaine placebo injections have been reported (Cascella et al. 1989; Muntaner et al. 1989), although the precise autonomic mechanisms were not known. Second, these responses indicated that a decrease in vagal tone is not a necessary correlate of tachycardia. Therefore, it appears unlikely that changes in vagal tone are artifactually related to changes in heart rate.
One might expect that this anticipatory heart rate response could be due to nonspecific orienting rather than to specific anticipation of cocaine. However, the orienting response is typically deceleratory (Jennings 1986) rather than the acceleration in heart rate that was found when the physician entered the room. Moreover, the orienting response has a significant vagal component (Obrist 1981). We did not find any concomitant change in vagal tone when the physician entered the subject chamber. Similar arguments can be made concerning the response to placebo.
These results contradict the prevailing view that the cardiovascular response to cocaine is due to sympathetic (Billman 1990; Ritchie and Greene 1990) rather than parasympathetic mechanisms. In fact, cocaine is a relatively potent vagal blocker. The implications of these results for the cardiotoxicity of cocaine remain to be investigated. Evidence that tonic parasympathetic inhibitory control of the heart normally suppresses arrhythmiagenesis (Kamarck and Jennings 1991) may be directly relevant. Withdrawal of vagal tone is associated with several different cardiac arrhythmias (Corr and Gillis 1974; Goldstein et al. 1973; Kent et al. 1973; Myers et al. 1974). Therefore, the relatively pronounced release of vagal inhibition produced by moderate dosages of cocaine may play a direct etiologic role in overdose of this drug.
The results obtained with measures derived from Lorenz plots of successive R to R intervals provided supportive evidence. The Vagal Index, which was most sensitive to atropine in a validation study (Toichi et al. 1997), was significantly decreased by i.v. cocaine. This effect was congruent with results using Porges’ (1995) Vagal Tone measure. The Sympathetic Index (Toichi et al. 1997) was not significantly affected by i.v. cocaine, which supported our conclusion that the tachycardia was not sympathomimetic, but was instead due to withdrawal of vagal (i.e., parasympathetic) inhibition. We did not find the robust sympathetic effects that would be predicted by the assumption of sympathetic mediation of the autonomic effects of cocaine. Instead, these measures provided converging evidence that the autonomic mechanism of the tachycardia from cocaine was withdrawal of vagal inhibitory influences on the heart.
The effect of i.v. cocaine on Pincus et al.'s (1991) ApEn was robust. There were substantial increases in the regularity (i.e., decreases in complexity) of the R to R intervals after cocaine was administered. Pincus and Goldberger (1994) have argued on the basis of empirical evidence that highly regular biological systems are characteristic of disease states, and that a “healthy” biological system is characterized by high complexity. Pincus (1994) speculated that high complexity represents the effects of multiple inputs to a biological system (such as the cardiovascular system), and that regularity of the system increases as these multiple inputs are reduced or eliminated. Therefore, the robust increase in regularity of heart period (i.e., reduction in ApEn) in the present study may represent a “disconnection” syndrome in which the impact of neural systems on the heart have been reduced (Pincus 1994). Certainly, the impact of parasympathetic inhibition was reduced by cocaine.
The subjects in this study were all men, and it is possible that different results would be obtained with women. We can only speculate on possible gender differences in the specific measures employed in this study since these measures have not been used in other cocaine research. The men in this study were polydrug abusers without significant cardiovascular problems or histories of adverse responses to cocaine. Although they were free of cocaine for at least a week before participating in the study, they could have been partially tolerant to the drug. New users, who are not tolerant to cocaine, may have even larger cardiovascular responses to the drug, and very recent users could have lesser effects due to tolerance. Finally, we recorded for 30 minutes following i.v. injections, and cardiovascular levels did not return fully to baseline levels during that time period. It is possible that different effects would obtain at later time points, although these could represent the beginning of acute withdrawal from cocaine rather than direct effects of the drug.
In conclusion, we developed a working hypothesis (Newlin 1995) that the cardiovascular effects of cocaine were due to withdrawal of vagal tone and to increased alpha-adrenergic activity, the latter affecting primarily peripheral resistance. The results of the present study tend to support the first part of our hypothesis, that cocaine reduces parasympathetic inhibition rather than increasing beta-adrenergic sympathetic activity.
References
Axelrod SD, Gordon D, Ubel FA, Shannon DC, Barger AC, Cohen RJ . (1981): Power spectrum analysis of heart rate fluctuations: A quantitative probe of beat-to-beat cardiovascular control. Science 213: 220–223
Ambre JJ, Belknap SM, Nelson J, Ruo TI, Shin SG, Atkinson AJ Jr . (1988): Acute tolerance to cocaine in humans. Clin Pharm Ther 44: 1–8
Berne RM, Levy MN . (1977): Cardiovascular Physiology, 3rd ed. St. Louis, Mosby
Billman GE . (1990): Mechanisms responsible for the cardiotoxic effects of cocaine. FASEB J 4: 2469–2475
Cascella N, Muntaner C, Kumor KM, Nagoshi CT, Jaffe JH, Sherer MA . (1989): Cardiovascular responses to cocaine placebo in humans: A preliminary report. Biol Psychiat 25: 285–295
Corr PB, Gillis RA . (1974): Role of the vagus nerves in the cardiovascular changes induced by coronary occlusion. Circulation 49: 86–97
Dellinger JA, Taylor HL, Porges SW . (1987): Atropine sulfate effects on aviator performance and on respiratory-heart period interactions. Aviat Space Environ Med 58: 333–338
Fischman MW, Schuster CR, Resnekov L, Shick JF, Krasnegor NA, Fennell W, Freedman DX . (1976): Cardiovascular and subjective effects of intravenous cocaine administration in humans. Arch Gen Psychiat 33: 983–989
Gantenberg NS, Hageman GR . (1991): Cocaine depresses cardiac sympathetic efferent activity in anesthetized dogs. J Cardiovas Pharmacol 17: 434–439
Goldstein RE, Karsh RB, Smith ER, Orlando M, Norman D, Farnham G, Epstein SE . (1973): Influence of atropine and of vagally mediated bradycardia on the occurrence of ventricular arrhythmias following acute coronary occlusion in closed-chest dogs. Circulation 47: 1180–1190
Jennings JR . (1986): Bodily changes during attending. In Coles M.G.H. Donchin E. Porges S.W. (eds) Psychophysiology: Systems, Processes, and Applications. New York, Guilford, pp 268–289
Johanson CE, Fischman MW . (1989): The pharmacology of cocaine related to its abuse. Pharmacol Rev 41: 3–52
Kamarck T, Jennings JR . (1991): Biobehavioral factors in sudden cardiac death. Psychol Bull 109: 42–75
Kent KM, Smith ER, Redwood DR, Epstein SE . (1973): Electrical stability of acutely ischemic myocardium: Influences of heart rate and vagal stimulation. Circulation 47: 291–298
London ED, Cascella NG, Wong DF, Phillips RL, Dannals RF, Links JM, Herning R, Grayson R, Jaffe JH, Wagner HN Jr . (1990): Cocaine-induced reduction in glucose utilization in human brain: A study using positron emission tomography and [fluorine 18]-fluorodeoxyglucose. Arch Gen Psychiat 47: 567–574
Muntaner C, Cascella NG, Kumor KM, Nagoshi C, Herning R, Jaffe J . (1989): Placebo responses to cocaine administration in humans: Effects of prior administrations and verbal instructions. Psychopharmacology 99: 282–286
Myers RW, Pearlman AS, Hyman RM, Goldstein RA, Kent KM, Goldstein RE, Epstein SE . (1974): Beneficial effects of vagal stimulation and bradycardia during experimental acute myocardial ischemia. Circulation 54: 943–947
Newlin DB, Pretorius MB . (1991): Prior exposures to the laboratory enhance the effect of alcohol. J Stud Alcohol 52: 470–473
Newlin DB . (1995): Effect of cocaine on vagal tone: A common factors approach. Drug Alcohol Depend 37: 211–216
Newlin DB, Pretorius MB, Wong CJ, Dax EM . (1991): Acute marijuana smoking reduces vagal tone. NIDA Research Monograph, Vol. 105. Committee on Problems of Drug Dependence, Washington, DC, US Gov Printing Office, pp 565–566
Newlin DB, Byrne EA, Porges SW . (1990): Vagal mediation of the effect of alcohol on heart rate. Alcoholism Clin Exp Res 14: 421–424
Obrist PA . (1981): Cardiovascular Psychophysiology. New York, Plenum
Pincus SM . (1994): Greater signal regularity may indicate increased system isolation. Math Biosci 122: 161–181
Pincus SM, Gladstone IM, Ehrenkranz RA . (1991): A regularity statistic for medical data analysis. J Clin Monit 7: 335–345
Pincus SM, Goldberger AL . (1994): Physiological time-series analysis: What does regularity quantify?. Am J Physiol 266: H1643–H1656
Porges SW, Bohrer RE . (1990): Analyses of periodic processes in psychophysiological research. In Cacciopo J.T. Tassihary L.G. Principles of Psychophysiology: Physical, Social, and Inferential Elements. New York, Cambridge University Press, pp 708–753
Porges SW . (1995): Method and Apparatus for Evaluating Rhythmic Oscillations in Aperiodic Physiological Response Systems. U.S. Patent No. 4,510,944
Pretorius MB, Wong CJ, Newlin DB . (1991): NIDA Research Monograph, Vol. 105 Committee on Problems of Drug Dependence, Washington, DC, US Gov Printing Office, pp 417–418
Raczkowski VF, Hernandez YM, Erzouki HK, Abrahams TP, Mandal AK, Hamosh P, Friedman E, Quest JA, Dretchen KL, Gillis RA . (1991): Cocaine acts in the central nervous system to inhibit sympathetic neural activity. J Pharmacol Exp Ther 257: 511–519
Rappolt RT, Gay GR, Inaba DS . (1977): Propranolol: A specific antagonist to cocaine. Clin Toxicol 10: 265–271
Resnick RB, Keslenbaum RS, Schwartz LK . (1977): Acute systemic effects of cocaine in man: A controlled study by intranasal and intravenous routes. Science 195: 696–698
Ritchie JM, Greene NM . (1990): Local anesthetics. In Gilman A.G. Rall T.W. Nies A.S. Taylor P. (eds), Goodman and Gilman's The Pharmacological Basis of Therapeutics 8th ed. New York, Pergamon, 311–331
Sharkey J, Ritz MC, Schenden JA, Hanson RC, Kuhar MJ . (1988): Cocaine inhibits muscarinic cholinergic receptors in heart and brain. J Pharm Exp Ther 246: 1048–1052
Tella SR, Schindler CW, Goldberg SR . (1990): The role of central and autonomic neural mechanisms in the cardiovascular effects of cocaine in conscious squirrel monkeys. J Pharmacol Exp Ther 252: 491–499
Toichi M, Sugiura T, Murai T, Sengoku A . (1997): A new method of assessing cardiac autonomic function and its comparison with spectral analysis and coefficient of variation of R-R interval. J Auton Nerv Syst 62: 79–84
Wilkerson RD . (1989): Cardiovascular effects of cocaine: Enhancement by yohimbine and atropine. J Pharmacol Exp Ther 248: 57–61
Wong CJ, Newlin DB, Pretorius MB, Better WB, Pickworth W . (1991): Acute effects of various abused drugs on heart rate and cardiac vagal tone: A common factors approach. Palm Beach, FL, Committee on Problems of Drug Dependence
Acknowledgements
A published report (Newlin 1995) from a College on Problems of Drug Dependence symposium presented in abstract form a limited subset of the results with vagal tone. The authors gratefully acknowledge the assistance of Merrily Smith, Valerie O'Brien, and Warren Better in the collection of this data. Roy Pickens, Michael Kuhar, Charles Schindler, and W. Robert Lange provided helpful comments on drafts of the manuscript. This article represents the views of the authors, and does not reflect those of NIDA or the U.S. Government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Newlin, D., Wong, C., Stapleton, J. et al. Intravenous Cocaine Decreases Cardiac Vagal Tone, Vagal Index (Derived in Lorenz Space), and Heart Period Complexity (Approximate Entropy) in Cocaine Abusers. Neuropsychopharmacol 23, 560–568 (2000). https://doi.org/10.1016/S0893-133X(00)00135-4
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1016/S0893-133X(00)00135-4
