
Abstract
Some studies have suggested that disorders in the peripheral and central metabolism of serotonin (5-HT) and noradrenaline (NE) may play roles in the pathophysiology of post-traumatic stress disorder (PTSD). This study examines (1) the availability of plasma total tryptophan, the precursor of 5-HT, and tyrosine, the precursor of NE; and (2) the platelet 5-HT transporter and α2-adrenoceptor (α2-AR) binding sites in patients with PTSD and healthy volunteers. High-performance liquid chromatography (HPLC) was employed to measure plasma tryptophan and tyrosine as well as amino acids known to compete with the same cerebral transport system; that is, valine, leucine, phenylalanine, and isoleucine. The maximum number of binding sites (Bmax) and their affinity (Kd) for binding to [3H]-paroxetine and [3H]-rauwolscine, a selective α2-AR antagonist, were determined. [3H]-paroxetine and [3H]-rauwolscine binding Kd values were significantly higher in patients with PTSD than in healthy volunteers. [3H]-rauwolscine binding Kd values were significantly higher in patients with PTSD and concurrent major depression (MD) than in PTSD patients without MD and healthy volunteers. Plasma tyrosine concentrations and the ratio of tyrosine/valine + leucine + isoleucine + phenylalanine + tryptophan were significantly higher in PTSD patients with MD than in those without MD and healthy volunteers. The results show that PTSD is accompanied by lower affinity of paroxetine binding sites and that PTSD with concurrent MD is accompanied by lower affinity of α2-ARs and increased plasma tyrosine availability to the brain. The results suggest that (1) serotonergic mechanisms, such as defects in the 5-HT transporter system, may play a role in the pathophysiology of PTSD; and (2) that catecholaminergic mechanisms, such as increased precursor availability and lowered affinity of α2-ARs, may play a role in the pathophysiology of PTSD with concurrent MD.
Similar content being viewed by others

Main
Post-traumatic stress disorder (PTSD) is an anxiety disorder that may occur after exposure to extreme traumatic events, such as natural disasters (e.g., flood), accidental (e.g., car accidents, fire), and deliberate man-made (e.g., war camps, torture, wars) traumatic events (APA 1980; 1987; 1994). The core symptoms and diagnostic criteria according to the DSM (III, III-R, and IV) are reexperiencing the trauma; numbing of responsiveness and avoidance; and hyperarousal. The condition is highly prevalent, with considerable morbidity and a high co-morbidity with other psychiatric disorders, such as major depression (APA 1980; 1987; 1994).
Recent research on the biological pathophysiology of PTSD has found some evidence for roles of catecholamines and serotonin (5-HT). Findings on increased catecholaminergic or sympathetic nervous system (SNS) activity in PTSD are fairly consistent across studies. First, combat veterans with PTSD show significantly higher 24-h urinary excretion and plasma concentrations of catecholamines, such as noradrenaline (NE), adrenaline, and dopamine, than normal controls, other psychiatric patients, or combat veterans without PTSD (Kosten et al. 1987; Mason et al. 1988; Yehuda et al. 1992; Hammer and Diamond 1993; Lemieux and Coe 1995; Yatham et al. 1996). Second, combat veterans show a significantly higher rise than normal volunteers in plasma NE and peripheral SNS activity following acute laboratory stressors with stimuli reminiscent of the trauma (Blanchard et al. 1991; Krystal et al. 1989). Third, PTSD patients have significantly decreased platelet [3H]-rauwolscine binding Bmax values, suggesting a downregulation of α2-adrenoceptors (α2-ARs) on platelets (Perry et al. 1987). Fourth, challenge with yohimbine, an α2-AR antagonist that preferentially blocks the presynaptic α2-AR autoreceptor (Starke et al. 1975), resulted in significantly higher plasma levels of 3-methoxy-4-hydroxyphenylglycol (MHPG), the major NE metabolite, in PTSD veterans than in control subjects (Southwick et al. 1993). These results suggest that central presynaptic α2-ARs are subsensitive in PTSD (Southwick et al. 1993). No significant alterations in 24-h urinary NE excretion and lower arterialized plasma NE concentrations have been found by other authors (Pitman and Orr 1990; Mellman et al. 1995; Murburg et al. 1995). However, to the best of our knowledge, no research has examined plasma tyrosine, the precursor of NE, in PTSD. There is evidence that the rate of formation of brain NE is determined by brain tyrosine concentrations, which are reflected in the plasma by the molar ratio of tyrosine to other amino acids known to compete for the same cerebral uptake site; that is, the competing amino acids (CAA) tryptophan, phenylalanine, leucine, isoleucine, and valine (reviews: Moller 1986a; Salter and Pogson 1987; Voog and Eriksson 1992). During stressful events, an increase in plasma tyrosine and the tyrosine/CAA is observed; whereas, the increased tyrosine/CAA ratio is related to increased brain noradrenergic turnover (review: Rao 1987).
Supportive findings that alternations in serotonergic metabolism may play roles in the pathophysiology of PTSD include the following. First, selective 5-HT reuptake inhibitors (SSRIs), such as fluoxetine, have beneficial effects in the treatment of PTSD (van der Kolk et al. 1994). Second, platelet [3H]-paroxetine binding Bmax and Kd values were significantly lower in PTSD patients than in normal controls, suggesting a lower number of platelet 5-HT transporters in PTSD patients (Arora et al. 1993). The 5-HT transporter plays a critical role in 5-HT neurotransmission by reclaiming synaptic 5-HT. Arora et al. (1993) have argued that platelet [3H]-paroxetine binding may reflect 5-HT transporter activity. Third, maximal responders to subchronic treatment with fluoxetine had significantly lower pretreatment platelet [3H]-paroxetine binding Kd values than partial responders, suggesting that lower Kd values are possible predictors of SSRI response in PTSD (Fichtner et al. 1994). However, whether PTSD is accompanied by lower availability of plasma total tryptophan, the precursor of 5-HT, remains elusive. Total plasma tryptophan as well as the tryptophan/CAA ratio are indicators for the availability of tryptophan to the brain (Fernstrom 1984) and, hence, for 5-HT synthesis in the brain (Moir and Eccleston 1968). Lowered plasma tryptophan availability is thought to play a role in the serotonergic pathophysiology of major depression (Maes and Meltzer 1995).
The aims of the present study were to examine whether PTSD with or without co-morbid major depression is accompanied by (1) serotonergic disorders, such as lowered platelet [3H]-paroxetine binding Bmax values, increased [3H]-paroxetine binding Kd values, and lower plasma tryptophan concentrations; and (2) noradrenergic disorders, such as lowered platelet [3H]-rauwolscine binding Bmax values, increased [3H]-rauwolscine binding Kd values, and increased plasma tyrosine concentrations.
SUBJECTS
In the present study, 57 subjects participated. In study group 1, we examined the plasma amino acids in 32 normal volunteers and 13 outpatients with PTSD. In study group 2, we examined platelet [3H]-paroxetine and [3H]-rauwolscine binding characteristics in 12 normal volunteers and 12 of the above patients with PTSD. The patients were victims of two accidental man-made traumatic events that occurred in Vlaanderen (i.e., a December 31, 1994 fire in an Antwerp hotel and a February 27, 1996 multiple collision, car crash, Deinze). The patients developed PTSD 9 months after the traumatic event. We employed the Composite International Diagnostic Interview (CIDI), PTSD Module, to make the diagnosis of PTSD according to the DSM-III-R criteria (Janca et al. 1994; Peters et al. 1996). The CIDI–PTSD module, is a structured diagnostic interview that covers all DSM-III-R (APA 1987) criteria for PTSD. Because we were unable to validate the DSM-III-R diagnostic criteria for PTSD in factor and cluster analytic studies performed on 185 victims from the above traumatic events, we reclassified the patients according to results of cluster analysis, which divided the 185 victims in cases and noncases (Maes et al. 1998a; 1998b). This cluster analytically derived classification was less conservative than that of the DSM-III-R. Of the 13 PTSD patients included here, nine had PTSD according to DSM-III-R criteria, and four patients were classified as being PTSD cases according to the cluster analysis.
Normal volunteers and PTSD patients had the usual blood tests (serum electrolytes, urea), liver function tests (γGT, SGPT, SGOT), and thyroid function tests (FT4 and basal thyroid secreting hormone). All subjects were free of any infections, and inflammatory or allergic reactions for at least 2 weeks before blood samplings. Exclusion criteria for PTSD patients and healthy volunteers were: subjects suffering from a neurological, inflammatory, endocrine or clinically significant chronic disease; immunocompromised subjects; subjects receiving drugs with known or potential interaction with plasma amino-acids, 5-HT and α2-ARs, and immune and endocrine functions. All healthy volunteers had a negative past, present, or family history for psychiatric disorders. None was a regular drinker and none had ever taken psychotropic drugs. All were free of any medications for at least 1 month.
Exclusion criteria for PTSD patients were a present or past history of other axis-I diagnoses, such as bipolar, schizophrenic, paranoid, organic mental, psychoactive substance use and eating disorders. Patients who had suffered from major depression before the traumatic event were also excluded. However, PTSD patients who developed major depression within 9 months after the trauma were included in the present study. Axis-I diagnoses were made according to DSM-III-R criteria (APA 1987), using the Structured Interview for the DSM-III-R, patient version (Spitzer et al. 1990). Severity of depression was measured by means of the Hamilton Depression Rating Scale (HDRS, Hamilton 1960). All PTSD patients with or without major depression were free of any psychotropic drugs and substance abuse since the traumatic event.
METHODS
Blood collections and clinical assessments were carried out 7–9 months after the traumatic event. Blood samples for the assays of plasma amino acids and platelet [3H]-paroxetine and [3H]-rauwolscine binding assays were collected at 7:45 A.M. (±15 min) after an overnight fast. All assays were done blind to the subject status. Plasma samples for the determination of the amino acids were kept at −75°C until thawed for assay. The amino acids were determined by means of a high-performance liquid chromatography (HPLC) method, as described previously (Turnell and Cooper 1982; Maes et al. 1996). All blood specimens for the assay of plasma amino acids in PTSD patients and healthy volunteers were assayed in a single run with a single lot number of reagents and consumables employed by a single operator (AL). The intra-assay coefficients of variation (CV) values obtained in our laboratory are: tryptophan 3.3%; tyrosine 3.8%; valine 3.0%; phenylalanine 3.2%; isoleucine 3.4%; and leucine 3.7%. The tryptophan/valine + leucine + isoleucine + tyrosine + phenylalanine (CAA1) and the tyrosine/valine + leucine + isoleucine + tryptophan + phenylalanine (CAA2) ratios were computed and multiplied by 100.
For platelet [3H]-paroxetine and [3H]-rauwolscine binding assays, 2 × 9 ml of blood was collected in silliconated tubes containing K-EDTA as anticoagulant. Within 60 min, platelet-rich plasma was prepared by low-speed centrifugation for 10 min at 1500 rpm (750g). Platelets were isolated in four centrifugation runs, pooled, counted, and washed in 8 ml buffer A (Tris 50 mm, NaCl 150 mm, EDTA 20 mm, pH 7.5). Platelets were counted by means of a Coulter STKS fully automated total blood cell counter. The final pellet was disrupted with and frozen in 2 ml ice-cold buffer B (Tris 5 mm, EDTA 5 mm, pH 7.5). Samples were frozen at −80°C until thawed for determination of binding assays. After thawing and adding 6 ml buffer B, the platelet membranes were centrifuged twice in a Beckman PG centrifuge at 18,000 rpm (39,000 g) and twice disrupted using an ultraturrax homogenizer. The final membrane pellet was suspended in 0.8 ml buffer C (glycylglycine 25 mm, pH 7.6), resulting in a final concentration of 4,000 × 10E6/ml.
For the [3H]-paroxetine binding assays, incubation mixtures consisted of 50 μl platelet membrane suspension, 400 μl buffer D (Tris HCl 50 mm, NaCl 120 mm, KCl 5mm), 25 μL [3H]-paroxetine at final concentrations between 0.02 and 0.8 nm, and 25 μl ethanol 10% or 25 μl imipramine (diluted in ethanol 10%, in a final concentration of 10 μm in incubation mixture; 1,000 × excess of [3H]-paroxetine). Specific binding (as a percentage of total binding) was 73 ± 5%. After incubation for 120 min at 25°C, samples were rapidly filtered through Whatman GF/B glass fiber filters under vacuum and rinsed twice with 4 ml ice-cold buffer. We used a 40-well filtration manifold. Filters were placed in vials with 2 ml scintillation fluid (Packard Ultima-Gold) and radioactivity was counted after 12 h of incubation in a Packard 460 scintillation counter. For the [3H]-rauwolscine binding assays, incubation mixtures, consisted of 50 μl platelet suspension, 50 μl 3[H]-rauwolscine at final concentrations between 0.1 and 4.0 nm and 400 μl buffer C. Specific binding was defined as radioactivity displaced by 10 μm idazoxan. Specific binding was 80 to 90%. The solution was incubated for 60 min at 25°C. The samples were rapidly filtered through Whatman GF/B glass fiber filters under vacuum and rinsed twice with 4 ml ice-cold buffer E (Tris 50 mm, pH 7.4). Filters were placed in vials with 2 ml scintillation cocktail (Packard Ultima-Gold). Radioactivity was counted after 16 h in a Packard 460 scintillation counter. Data were analyzed using computerized curve fitting; that is, LIGAND. Bmax, and Kd values were derived from the calculated curves. Bmax was expressed as fmoles/10E6 platelets. Blood specimens for the assay of platelet [3H]-paroxetine and [3H]-rauwolscine binding characteristics were carried out in three assays on 3 different days with a single lot number of reagents and consumables employed by a single operator (LD). To minimize the interassay analytical variability, we analyzed four samples of PTSD patients and four age–sex matched controls in each of the three assays and minimized possible effects of interassay variability on our results by means of regression analysis.
Severity of PTSD was assessed as described previously (Maes et al. 1998a; 1998b). In brief, a factor analysis (principal component method) was performed on all CIDI items in 185 victims of the above traumatic events. The factor scores on the first two varimax-rotated factors were used as measures for the severity of two different dimensions. The first factor, “depression-avoidance (DAV) dimension,” contains the following items: restricted range of affect; markedly diminished interest; feelings of detachment; sense of a foreshortened future; inability to recall an important aspect of the trauma; acting or feeling as if the traumatic event is recurring; and effects to avoid activities, places, or people that arouse recollections of the trauma. The second factor, “arousal–anxiety (AA) dimension,” contains the following items: physiological symptoms of anxiety; sleep disorders; recurrent distressing dreams; recurrent and intrusive distressing recollections of the event; effects to avoid thoughts, feelings, or conversations associated with the trauma; hypervigilance; intense physiological distress at exposure to cues that symbolize the traumatic event; exaggerated startle response.
STATISTICS
The independence between classification systems was checked with the analysis of contingence (χ2 test). Group mean differences were checked by means of analysis of variance (ANOVA) or analysis of covariance (ANCOVA). Multiple post hoc differences between group means were checked with Fisher's least significant difference (LSD). Relationships between variables were assessed with Pearson's product-moment correlation coefficients and through multiple regression analyses. Normality of distribution was assessed with the Kolmogorov–Smirnov test. Transformations were used to reach normality of distribution or to adjust for heterogeneity of variance between study groups; that is, platelet [3H]-paroxetine and [3H]-rauwolscine binding values were processed after square root transformation.
RESULTS
Demographic data
In study group 1, there were no significant differences in age (F = 0.4, df = 1/43, p = .3) or gender (χ2 = 1.3, df = 1, p = .2) between patients with PTSD (mean age = 47.1 ± 9.3 years; M/F ratio = 4/9) and healthy volunteers (mean age: 45.3 ± 7.7 years; M/F ratio = 5/27). In study group 2, there were no significant differences in age (F = 0.5, df = 1/22, p = .5) or gender (χ2 = 0.0, df = 1, p = 1.0) between patients with PTSD (mean age = 49.1 ± 9.0 years; M/F ratio = 4/8) and healthy volunteers (mean age = 46.1 ± 10.8 years; M/F ratio = 4/8). There were no significant correlations between age and plasma tryptophan (r = −0.28, p = .95); tyrosine (r = 0.00, p = .98); tryptophan/CAA1 ratio (r = −0.01, p = .95); [3H]-paroxetine binding Bmax (r = 0.30, p = .1) or Kd (r = 0.17, p = .6) values; and [3H]-rauwolscine binding Bmax (r = −0.22, p = .3) or Kd (r = 0.30, p = .2) values. There was a significant and positive correlation between age and the tyrosine/CAA2 ratio (r = 0.36, p = .01) (all results of intraclass correlations, pooled over the study groups of normal controls and PTSD patients). ANOVAs, factorial design with gender as second factor, showed no significant differences between males and females in plasma tryptophan (F = 2.4, df = 1/41, p = .1); tryptophan/CAA1 (F = 3.7, df = 1/41, p = .06); tyrosine/CAA2 (F = 0.7, df = 1/41, p = .6); [3H]-paroxetine binding Bmax (F = 1.0, df = 1/20, p = .3) or Kd (F = 0.0, df = 1/20, p = .8) values; and [3H]-rauwolscine binding Bmax (F = 1.3, df = 1/20, p = .3) or Kd (F = 0.0, df = 1/20, p = .9) values. Tyrosine was significantly higher in males (mean = 70 ± 8 μmol/l) than in females (mean = 57 ± 11 μmol/l) (F = 6.3, df = 1/41, p = .01). There were no significant differences in the [3H]-paroxetine binding Bmax values (F = 2.7, df = 2/21, p = .09) between the three assays. There were significant differences in [3H]-paroxetine binding Kd (F = 4.4, df = 2/21, p = .02); [3H]-rauwolscine binding Bmax (F = 4.5, df = 2/21, p = .02) and Kd (F = 5.6, df = 2/21, p = .01) values between the three assays. In any case, we have adjusted subsequent statistical analyses for possible age and gender effects and analytical interassay variability by entering age, sex, and the three analytical assays (entered as two dummy variables) as covariates in regression analyses. At the time of blood sampling, seven patients suffered from PTSD without MD and six from PTSD with MD. The mean HDRS (±SD) score of patients with co-morbid depression and PTSD (19.2 ± 3.4) was significantly higher than that of patients with PTSD only (7.3 ± 2.2) (F = 51, df = 1/10, p = .0001).
Biological markers and PTSD
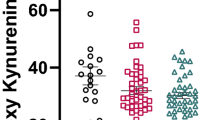
Table 1 shows that plasma tyrosine and the tyrosine/CAA2 ratio were significantly higher in PTSD subjects than in normal volunteers. ANCOVAs performed on PTSD patients divided into those with and without MD, showed that plasma tyrosine and the tyrosine/CAA2 ratio were significantly higher in PTSD patients with MD than in normal volunteers and PTSD patients without MD. There were no significant differences in plasma tryptophan, tryptophan/CAA1 ratio, CAA1 or CAA2 between PTSD patients and healthy volunteers.
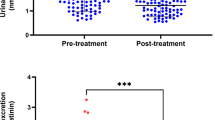
Figure 1 shows that the [3H]-paroxetine binding Kd values were significantly higher (F = 8.2, df = 1/18, p = .009) in the PTSD patients (mean = 0.048 ± 0.016 nm), irrespective of the presence of MD, than in healthy volunteers (mean = 0.032 ± 0.012 nm). There were no significant differences between PTSD patients with and without MD. Figure 2 shows that the [3H]-rauwolscine binding Kd values were significantly higher (F = 5.0, df = 1/18, p = .04) in PTSD patients (mean = 0.187 ± 0.090 nm) than in normal volunteers (mean = 0.138 ± 0.057 nm). PTSD patients with MD (mean = 0.234 ± 0.106 nm) had significantly higher (F = 4.4, df = 2/17, p = .03) [3H]-rauwolscine binding Kd values than normal volunteers and PTSD patients without MD (mean = 0.140 ± 0.036 nm). There were no significant differences (F = 0.9, df = 1/18, p = .6) in [3H]-paroxetine binding Bmax values between PTSD patients (mean = 0.372 ± 0.181 fmoles/106 platelets) and healthy volunteers (mean = 0.302 ± 0.090 fmoles/106 platelets). There were no significant differences (F = 1.8, df = 1/18, p = .6) in [3H]-rauwolscine binding Bmax values between PTSD patients (mean = 0.113 ± 0.050 fmoles/106 platelets) and healthy volunteers (mean = 0.092 ± 0.030 fmoles/106 platelets) (all results of ANCOVAs with the analytical assays and/or age and gender as co-variates; and of LSD performed after ANCOVAs).

[3H]-paroxetine binding Kd values in healthy volunteers (HV), and patients with post-traumatic stress disorder (PTSD) with (PTSD + MD) and without (PTSD-MD) major depression.

[3H]-rauwolscine binding Kd values in healthy volunteers (HV), and patients with post-traumatic stress disorder (PTSD) with (PTSD + MD) and without (PTSD-MD) major depression.
To confirm the above results in patients with DSM-III-R diagnosis of PTSD only, we performed additional ANCOVAS on the study groups of normal volunteers and the PTSD patients classified according to DSM-III-R criteria. Serum tyrosine was significantly higher (F = 9.7, df = 1/37, p = .004) in PTSD patients with a DSM-III-R PTSD diagnosis (mean = 69.64 ± 9.86 μmol/l) than in healthy volunteers (mean = 55.94 ± 10.70 μmol/l). The tyrosine/CAA2 ratio (×100) was significantly greater (F = 6.6, df = 1/37, p = .01) in PTSD patients with a DSM-III-R PTSD diagnosis (mean = 12.43 ± 1.99) than in healthy volunteers (mean = 10.91 ± 1.42). [3H]-paroxetine binding Kd values (F = 6.3, df = 1/14, p = .02) were significantly higher in the PTSD patients with a DSM-III-R PTSD diagnosis (mean = 0.051 ± 0.018 nm) than in normal volunteers (mean = 0.032 ± 0.012 nm). [3H]-rauwolscine binding Kd values (F = 5.3, df = 1/15, p = .03) were significantly higher in the PTSD patients with a DSM-III-R PTSD diagnosis (mean = 0.194 ± 0.112 nm) than in normal volunteers (mean = 0.138 ± 0.057 nm.
There was a significant and positive relationship between [3H]-paroxetine binding Kd values and the DAV (F = 8.1, p = .02) and AA (F = 6.2, p = .04) factor scores. There was a trend toward a positive relationship between [3H]-rauwolscine binding Kd and the DAV (F = 4.5, p = .07) and AA (F = 2.7, p = .1) scores. There were no significant relationships between plasma tyrosine, the tyrosine /CAA2 ratio, plasma tryptophan, the tryptophan/CAA1 ratio, or [3H]-rauwolscine binding Bmax values and the AA or DAV scores (all results of partial correlation correlations with the three analytical assays and/or age and sex as covariates). There was a positive and significant correlation between the HDRS score and the tyrosine/CAA2 ratio (r = 0.61, p = .03) and a trend toward a significant correlation with [3H]-rauwolscine binding Kd values (r = 0.45, p = .1)
DISCUSSION
This study showed that the affinity of platelet [3H]-paroxetine binding sites is significantly decreased in patients with PTSD, irrespective of the presence of major depression. The significant and positive correlations between severity of PTSD, either the avoidance-depression or arousal-anxiety symptoms, may suggest that graded differences in the expression of PTSD symptoms are related to changes in the 5-HT transporter system. Arora et al. (1993) reported significantly lower [3H]-paroxetine binding Bmax and Kd values in PTSD patients than in healthy volunteers and found a negative correlation between the [3H]-paroxetine binding Bmax values and a state-dependent anxiety score. Unlike patients with PTSD, no significant alterations in platelet [3H]-paroxetine binding Bmax or Kd values could be found in major depression (Galzin et al. 1988; Lawrence et al. 1990; D'Hondt et al. 1994). These negative findings in major depression are in agreement with our results that there were no significant differences in platelet [3H]-paroxetine binding characteristics between PTSD patients with and without major depression.
A question, however, is whether peripheral findings are relevant for central serotonergic activity. Blood platelets are able to take up, store, and release 5-HT via mechanisms that are very similar to those of central 5-HT neurons (Sneddon 1969; 1973; Stahl 1977). [3H]-paroxetine binds with high affinity to a specific population of binding sites located on platelets and neuronal membranes associated with 5-HT uptake mechanisms (Mellerup et al. 1983; Habert et al. 1985; Cheetham et al. 1993). It has been found that the cloned 5-HT transporter on human platelets is identical to the 5-HT transporter in human brain (Lesch et al. 1993). Finally, the findings of Fichtner et al. (1994), who found a positive response to fluoxetine pharmacotherapy in PTSD patients with lower platelet [3H]-paroxetine binding characteristics, suggest that findings on platelet [3H]-paroxetine binding may be relevant for central serotonergic activity. The findings above may suggest that PTSD is accompanied by defects in the peripheral and perhaps central 5-HT transporter system.
In the present study, we were unable to find any significant differences in the availability of plasma tryptophan between PTSD patients and healthy volunteers. In this respect, Mellman and Kumar (1994) were unable to find significant differences either in platelet 5-HT concentrations or in platelet 5-HT uptake (Km and Vmax) measures between PTSD patients and controls.
We found that PTSD with comorbid major depression is accompanied by alterations in the peripheral catecholaminergic system; that is, higher plasma availability of tyrosine and lower affinity of platelet α2-ARs. There is some evidence that stressors lead to increases in the peripheral plasma tyrosine/CAA ratio, which corresponds to increased brain NE turnover (Rao 1987). Possible differences in diet between PTSD patients and controls are not likely to be involved in the increased tyrosine/CAA ratio found in PTSD. Although consumption of high/low protein or carbohydrate meals may acutely affect the tyrosine/CAA ratio in serum, fasting serum tyrosine concentrations (and the tyrosine/CAA ratio) did not differ significantly between humans on a control and protein-free diet over 18 days (Weller et al. 1969; Moller 1986b). A more plausible explanation for our findings revolves around the effects of NE hyperactivity in PTSD. Indeed, NE administration to rats may increase plasma tyrosine concentrations through changes in hepatic blood flow, which are probably α-AR related (Eriksson and Carlsson 1982; Rao 1987). Interestingly, previous research has shown that supplementary dietary tyrosine may be useful therapeutically in persons chronically exposed to psychological stress (Lehnert et al. 1984; Owasoyo et al. 1992). Some, but not all, studies in depression reported a reduced availability of plasma tyrosine (review: Maes et al. 1994). These results may indicate a distinct biochemical profile with respect to plasma tyrosine in depressed patients, as compared to those with major depression and PTSD.
The findings of the present study show that the affinity of [3H]-rauwolscine binding to platelets is significantly lower in patients with PTSD with a concurrent major depression than in healthy volunteers and PTSD patients without major depression. The biological significance of increased Kd values obtained for antagonists may be questioned. However, inhibition of [3H]-dihydroergocryptine binding by antagonists showed that there was a single population of antagonist binding sites with the pharmacological characteristics of α2-ARs; whereas, inhibition by agonists showed two sites with different affinities (Hoffman et al. 1979). Moreover, Perry et al. (1987) reported decreased platelet [3H]-rauwolscine binding Bmax values in PTSD. Adrenoceptors of the α2A subtype are expressed on platelets and presynaptically on noradrenergic neurons (Hieble et al. 1995; Limberger et al. 1995). Because platelet α2-ARs have very similar pharmacologic and kinetic properties as those in the brain, platelet α2-ARs are an adequate model for brain α2-ARs (Stahl 1985). Subsensitivity of central α2-ARs in PTSD has been suggested by Southwick et al. (1993). No significant differences in growth hormone response to desmethylimipramine administration were found between PTSD patients and normal subjects, suggesting no significant changes in central postsynaptic α2-ARs in PTSD (Yatham et al. 1996).
There are some reports that major depression per se is accompanied by subsensitive platelet α2-ARs. This evidence includes the following: (1) reduced platelet α2-AR responsiveness as measured by means of agonist-induced platelet aggregation and NE-induced inhibition of prostaglandine E1 (PGEI)-stimulated cyclic AMP (cAMP) production (Kafka et al. 1986; Kafka and Paul 1986); and (2)the density or affinity of platelet α2-ARs, as measured by [3H]-yohimbine and [3H]-rauwolscine binding to platelets, is lower in depressed patients than in healthy volunteers (Mendlewicz et al. 1989; Maes et al. 1998c). Studies in depression using partial agonists as ligands; for example clonidine, para-aminoclonidine, or UK-14304, often reported higher α2-AR Bmax values (review: Katona et al. 1989). These compounds, however, may also bind with high affinity to catecholamine-insensitive sites; that is, imidazoline sites, which may be upregulated in major depression (Piletz and Halaris 1994). Taken together, platelet and central presynaptic autoinhibitory α2-ARs may be subsensitive in PTSD or a subgroup thereof; that is, those with concurrent major depression. In this respect, it has been hypothesized that hyposensitivity of presynaptic α2-ARs may underlie increased SNS activity (see introductory section) and exaggerated plasma NE/MHPG response to various challengers in major depression (Potter and Manji 1994, Maes et al. 1998c) and PTSD (Southwick et al. 1993; Grillon et al. 1996).
It should be underscored, however, that the subjects being studied here are not typical of the kind of PTSD patients that have been studied in other reports. Indeed, most previous studies have examined patients who are symptomatic for decades; whereas, we studied PTSD patients within 9 months after the traumatic event. Thus, differences between studies may be present as a function of time elapsed since the trauma.
In conclusion, the results suggest that serotonergic mechanisms, such as a defect in the 5-HT transporter system, may play a role in the pathophysiology of PTSD. Our results of increased plasma tyrosine availability and lowered affinity of platelet α2-ARs are in accordance with previous findings that PTSD is accompanied by increased SNS activity but suggest that the latter may be confined to patients with co-morbid PTSD and major depression.
References
American Psychiatric Association (APA). (1980): Diagnostic and Statistical Manual of Mental Disorders, 3rd ed, revised. Washington, DC, APA
American Psychiatric Association (APA). (1987): Diagnostic and Statistical Manual of Mental Disorders, 3rd ed, revised. Washington, DC, APA
American Psychiatric Association (APA). (1994): Diagnostic and Statistical Manual of Mental Disorders, 4th ed, revised. Washington, DC, APA
Arora RC, Fichtner CG, O'Connor F, Crayton JW . (1993): Paroxetine binding in the blood platelets of post-traumatic stress disorder patients. Life Sci 53: 919–928
Blanchard EB, Kolb LC, Prins A, Gates S, McCoy GC . (1991): Changes in plasma norepinephrine to combat-related stimuli among Vietnam veterans with post traumatic stress disorder. J Nerv Ment Disord 179: 371–373
Cheetham SC, Viggers JA, Slater NA, Heal DJ, Buckett WR . (1993): [3H]Paroxetine binding in rat frontal cortex strongly correlates with [3H]-5-HT uptake: Effect of administration of various antidepressant treatments. Neuropharmacol 32: 737–743
D'Hondt P, Maes M, Leysen J, Gommeren W, Scharpé S, Cosyns P . (1994): Binding of [3H]-paroxetine to platelets of depressed patients: Seasonal differences and effects of diagnostic classification. J Affect Disord 32: 27–35
Eriksson T, Carlsson A . (1982): Adrenergic influence on rat plasma concentration of tyrosine and tryptophan. Life Sci 30: 1465–1472
Fernstrom JD . (1984): Tryptophan availability and serotonin synthesis in rat brain: Effects of experimental diabetes. In Schlossberger HG, Kochen W, Linzen B, Steinhart H (eds), Progress in Tryptophan and Serotonin Research. Berlin-New York, Walter de Gruyter, pp 161–172
Fichtner CG, Arora RC, O'Connor FL, Crayton JW . (1994): Platelet paroxetine binding and fluoxetine pharmacotherapy in post-traumatic stress disorder: Preliminary observations on a possible predictor of clinical treatment response. Life Sci 54: 39–44
Galzin AM, Poirier M-F, Loo H, Sechter D, Zarifian E, Langer SZ . (1988): [3H]-Paroxetine binding from healthy volunteers and depressed patients. Br J Pharmacol 93: 12
Grillon C, Southwick SM, Charney DS . (1996): The psychobiological basis of post-traumatic stress disorder. Mol Psychiatr 1: 278–297
Habert E, Graham D, Tahraoui L, Claustre Y, Langer S . (1985): Characterization of [3H]-paroxetine binding to rat cortical membranes. Eur J Pharmacol 118: 107–114
Hamilton M . (1960): A rating scale for depression. J Neurol Neurosurg Psychiatr 23: 56–61
Hammer MB, Diamond BI . (1993): Elevated plasma dopamine in post-traumatic stress disorder: A preliminary report. Biol Psychiatr 33: 304–306
Hieble JP, Bondinell WE, Ruffolo RR . (1995): α- and β-adrenoceptors: From the gene to the clinic. 1. Molecular biology and adrenoceptor classification. J Med Chem 38: 3415–3444
Hoffman BB, DeLean A, Wood CL, Schocken DD, Lefkowitz RJ . (1979): Alpha-adrenergic subtypes: Quantitative assessment by ligand binding. Life Sci 24: 1739–1746
Janca A, Üstun TB, Sartorius N . (1994): New versions of World Health Organization instruments for the assessment of mental disorders. Acta Psychiatr Scand 90: 73–83
Kafka MS, Paul SM . (1986): Platelet α2-adrenergic receptors in depression. Arch Gen Psychiatr 43: 91–95
Kafka MS, Nurnberger JI, Siever L, Targum S, Uhde TW, Gershon ES . (1986): Alpha-2 adrenergic receptor function in patients with unipolar and bipolar affective disorders. J Affect Disord 10: 163–169
Katona CLE, Theodorou AE, Davies SL, Hale AS, Kerry SM, Horton RW, Kelly JS, Paykel ES . (1989): [3H]-Yohimbine binding to platelet α2-adrenoceptors in depression. J Affect Disord 17: 219–228
Kosten TR, Mason JW, Giller EL, Ostroff RB, Harkness L . (1987): Sustained urinary norepinephrine and epinephrine elevation in post-traumatic stress disorder. Psychoneuroendocrinology 12: 13–20
Krystal JH, Kosten TR, Perry BD, Southwick SM, Mason JW, Giller EL . (1989): Neurobiological aspects of PTSD: Review of clinical and preclinical studies. Behav Ther 20: 177–198
Lawrence KM, De Paermentier F, Cheetham SC, Crompton MR, Katona CL, Horton RW . (1990): Brain 5-HT uptake sites, labeled with [3H]-paroxetine, in antidepressantfree depressed suicides. Brain Res 526: 17–22
Lehnert H, Reinstein DK, Strowbridge BW, Wurtman RJ . (1984): Neurochemical and behavioral consequences of acute, uncontrollable stress: Effects of dietary tyrosine. Brain Res 303: 215–223
Lemieux AM, Coe CL . (1995): Abuse-related post-traumatic stress disorder: Evidence for chronic neuroendocrine activation in women. Psychosom Med 57: 105–115
Lesch KP, Wolozin BL, Murphy DL, Reiderer P . (1993): Primary structure of the human platelet serotonin uptake site: Identity with the brain serotonin transporter. J Neurochem 60: 2319–2322
Limberger N, Funk L, Trendelenburg AU, Starke K . (1995): Subclassification of presynaptic α2-adrenoceptors: α2-autoreceptors in rabbit atria and kidney. Naunyn-Schmiedeberg's Arch Pharmacol 352: 31–42
Maes M, Meltzer HY, Cosyns P, Schotte C . (1994): Evidence for the existence of major depression with and without anxiety features. Psychopathology 27: 1–13
Maes M, Meltzer HYM . (1995): The serotonin hypothesis of major depression. In Bloom FE, Kupfer DJ (eds), Psychopharmacology, the Fourth Generation of Progress. New York, Raven Press, pp 933–944
Maes M, Wauters A, Verkerk R, Neels H, vanGastel A, Cosyns P, Scharpé S, Desnyder R . (1996): Lower L-tryptophan availability in depression: A marker of a more generalized disorder in protein metabolism. Neuropsychopharmacology 15: 243–251
Maes M, Delmeire L, Schotte C, Janca A . (1998a): Epidemiologic and phenomenological aspects of post-traumatic stress disorder: DSM-III-R diagnosis and diagnostic criteria not validated. Psychiatr Res, in press.
Maes M, Delmeire L, Creten T, Janca A . (1998b): The two-factorial symptom structure of post-traumatic stress disorder: Depression-avoidance and arousal-anxiety. Psychiatr Res, in press.
Maes M, Van Gastel A, Delmeire L, Meltzer HY . (1998c): Decreased platelet alpha-2 adrenoceptor density in major depression: Effects of antidepressive drugs. Biol Psychiatr, in press.
Mason JW, Giller EL, Harkness L . (1988): Elevation of urinary norepinephrine/cortisol ratio in post-traumatic stress disorder. J Nerv Ment Disord 176: 498–502
Mellerup ET, Plenge P, Engelstoft M . (1983): High affinity binding of [3H]-paroxetine and [3H]-imipramine to human platelet membranes. Eur J Pharmacol 96: 303–309
Mellman TA, Kumar AM . (1994): Platelet serotonin measures in post-traumatic stress disorder. Psychiatr Res 53: 99–101
Mellman TA, Kumar A, Kulick-Bell R, Kumar M, Nolan B . (1995): Nocturnal/daytime urine noradrenergic measures and sleep in combat related PTSD. Biol Psychiatr 38: 174–179
Mendlewicz J, Hirsch D, Sevy S, Surmont D, Papadimitriou G, De Maertelaer V . (1989): Alpha-2-adrenoceptor binding as a possible vulnerability marker for affective disorder. Neuropsychobiology 22: 61–67
Moir ATB, Eccleston D . (1968): The effect of precursor loading in the cerebral metabolism of 5-hydroxyindoles. J Neurochem 15: 1093–1108
Moller SE . (1986a): Plasma neutral amino acids and food preferences: Possible implications in normals and depressives. Biblthca Nutr Dieta 38: 149–153
Moller SE . (1986b): Carbohydrate/protein selection in a single meal correlated with plasma tryptophan and tyrosine ratios to neutral amino acids in fasting individuals. Physiol Beh 38: 175–183
Murburg MM, McFall ME, Lewis N, Veith RC . (1995): Plasma norepinephrine kinetics in patients with post-traumatic stress disorder. Biol Psychiatr 38: 819–825
Owasoyo JO, Neri DF, Lamberth JG . (1992): Tyrosine and its potential use as a countermeasure to performance decrement in military sustained operations. Aviat Space Environ Med 63: 364–369
Perry BD, Giller EL, Southwick SM . (1987): Altered platelet alpha 2-adrenergic binding sites in post-traumatic stress disorder. Am J Psychiatr 144: 1511–1512
Peters L, Andrews G, Cottler LB, Chatter S, Janca A, Smeets RMW . (1996): The composite International Diagnostic interview post-traumatic stress disorder module: Preliminary data. Int J Meth Psychiatr Res 6: 167–174
Piletz JE, Halaris A . (1994): Psychopharmacology of imidazoline and α2-adrenergic receptors: Implications for depression. Crit Rev Neurobiol 9: 29–66
Pitman RK, Orr SP . (1990): Twenty-four hour urinary cortisol and catecholamine excretion in combat-related post-traumatic stress disorder. Biol Psychiatr 27: 245–247
Potter WZ, Manji HK . (1994): Catecholamines in depression: An update. Clin Chem 40: 279–287
Rao M-L . (1987): Bioavailability of amino acids and amino acid precursors for neurotransmitter action: The role of hormones. In Huether G (ed), Amino Acid Availability and Brain Function in Health and Disease. Berlin, Springer-Verlag, pp 29–37
Salter M, Pogson CI . (1987): The importance of the liver as a regulator of amino acid supply to the brain. In Huether G (ed), Amino Acid Availability and Brain Function in Health and Disease. Berlin, Springer-Verlag, pp 21–28
Sneddon JM . (1969): Sodium-dependent accumulation of 5-hydroxytryptamine by rat blood platelets. Br J Pharmacol 37: 680–688
Sneddon JM . (1973): Blood platelets as a model for monoamine-containing neurons Progr Neurobiol 1: 151–198
Southwick SM, Krystal JH, Morgan CA, Johnson D, Nagy LM, Nicolaou A, Heninger GR, Charney DS . (1993): Abnormal noradernergic function in post-traumatic stress disorder. Arch Gen Psychiatr 50: 226–274
Spitzer RL, Williams JBW, Gibbon MSW, First MB . (1990): Structured Clinical Interview according to DSM-III-R. Washington, DC, American Psychiatric Press
Stahl SM . (1977): The human platelet: A diagnostic and research tool for the study of biogenic amines in psychiatry and neurologic disorders. Arch Gen Psychiatr 34: 509–516
Stahl SM . (1985): Peripheral models for the study of neurotransmitter receptors in man. Psychopharmacol Bull 21: 663–671
Starke K, Borowski E, Endo T . (1975): Preferential blockade of presynaptic α-adrenoceptors by yohimbine. Eur J Pharmacol 34: 385–388
Turnell D, Cooper J . (1982): Rapid assay for amino acids in serum or urine by precolumn derivatisation and reversed-phase liquid chromatography. Clin Chem 28: 527–531
van der Kolk BA, Dreyfuss D, Michaels M, Shera D, Berkowitz R, Fisler R, Saxe G . (1994): Fluoxetine in post-traumatic stress disorder. J Clin Psychiatr 55: 517–522
Voog L, Eriksson T . (1992): Relationship between plasma and brain large neutral amino acids in rats fed diets with different compositions at different times of the day. J Neurochem 59: 1868–1874
Weller LA, Margen S, Calloway DH . (1969): Variation in fasting and postprandial amino acids of men fed adequate or protein-free diets. Am J Clin Nutr 22: 1577–1583
Yatham LN, Sacamano J, Kusumakar V . (1996): Assessment of noradrenergic functioning in patients with noncombat-related post-traumatic stress disorder: A study with desmethylimipramine and orthostatic challenges. Psychiatr Res 63: 1–6
Yehuda R, Southwick S, Giller EL, Ma X, Mason JW . (1992): Urinary catecholamine excretion and severity of PTSD symptoms in Vietnam combat veterans. J Nerv Ment Disord 180: 321–325
Acknowledgements
This research was supported in part by the Fund for Scientific Research, Vlaanderen (FWO); the Clinical Research Center for Mental Health (CRC-MH), Antwerp; and Pfizer, Belgium. The assistance of T. Creten, E. DeBoel, G. Belis, CRC-MH, Antwerp; Prof. J. Mylle, Royal Military School, Brussels; Prof. B. VanHoudenhove, M.D., KUL, Leuven; Prof. Dr. J. Leysen and W. Gommeren, the Janssen Research Foundation, Beerse, Belgium; and Assoc. Prof. C. S. North, M.D., Washington University, St. Louis, USA is greatly appreciated. The authors thank Slachtofferhulp Vlaanderen; the “Stad Antwerpen” (L. Detiege); the O.C.M.W. Antwerpen; Politie van Antwerpen and Slachtofferzorg (B. Engelen, R. VanLooveren); Slachtofferhulp, Antwerpen; the Red Cross, Vlaanderen; and the “Dienst Dringende Spoed Interventie” (D. DeBeukelaer, H. Van Gastel). The secretarial assistance of Ms M. Maes is greatly appreciated.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Maes, M., Lin, Ah., Verkerk, R. et al. Serotonergic and Noradrenergic Markers of Post-Traumatic Stress Disorder with and without Major Depression. Neuropsychopharmacol 20, 188–197 (1999). https://doi.org/10.1016/S0893-133X(98)00058-X
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1016/S0893-133X(98)00058-X
Keywords
This article is cited by
-
Escitalopram reversed the traumatic stress-induced depressed and anxiety-like symptoms but not the deficits of fear memory
Psychopharmacology (2016)
-
Modulation of early stress-induced neurobiological changes: a review of behavioural and pharmacological interventions in animal models
Translational Psychiatry (2014)
-
Posttraumatic Stress Disorder and Comorbidity: Untangling the Gordian Knot
Psychological Injury and Law (2014)
-
Biological studies of post-traumatic stress disorder
Nature Reviews Neuroscience (2012)
-
Posttraumatic Stress Disorder in Children and Adolescents: A Review of Psychopharmacological Treatment
Child Psychiatry & Human Development (2010)
