
Abstract
The frequency of carriers of the AGUFin mutation, the predominant mutation causing aspartylglucosaminuria in Finland, was determined in a population sample comprising 553 newborns from a delivery hospital in southern Finland, and 607 from a hospital in northern Finland. The AGUFin point mutation was identified from cord blood samples using the PCR-based, solid-phase minisequencing method. Nineteen carriers of the AGUFin mutation were detected, 8 (1:69) in the sample from the southern and 11 (1:55) from the northern population, respectively. The solid-phase minisequencing method proved to be rapid and convenient for the detection of the AGUFin mutation, and can readily be applied in large-scale carrier screening at the population level.
Similar content being viewed by others
Introduction
Aspartylglucosaminuria (AGU) is a recessively inherited lysosomal storage disease, enriched in the Finnish population [1]. The disease is caused by deficient activity of aspartylglucosaminidase (AGA) resulting in the accumulation of glycoasparagines in the lysosomes [2]. AGU is clinically characterized by severe mental retardation, susceptibility to infections and various connective tissue manifestations. The first signs of the disease present between 1 and 4 years of age usually as a delay in speech and/or motor development. At the adult age, AGU patients are severely mentally retarded with characteristic coarse facial features. Their life span is shortened to 30–40 years [3].
Over 200 AGU patients have been identified in Finland in contrast to approximately 40 cases in the rest of the world. The disease is somewhat more frequent in northern and eastern Finland, the estimations of the carrier frequency ranging from 1:30 to 1:80 based on the prevalence of the disease [3, 4]. So far the heterozygotes have been identified by reduced AGA activity in lymphocytes [5]. However, the high individual variation in AGA activity and the laborious technique make the enzyme assay less suitable for large-scale carrier screening.
The mutation responsible for the AGU disease in Finland has recently been identified. The predominant mutation (AGUFin) is a point mutation (G > C) at base pair position 488 of the AGA cDNA, causing a cysteine to serine substitution at amino acid position 163 of the AGA protein. The Cys163 → Ser mutation is always linked to another point mutation only 5 bp apart which changes arginine at position 161 to glutamine. This mutation does not influence the enzyme activity, and its biological significance is still unknown [6]. Ninety-eight percent of the AGU alleles in the Finnish population carry these two mutations [7]. So far, only 5 Finnish compound heterozygous AGU patients have been identified with the AGUFin mutation in one allele and another yet unknown mutation in the other [7].
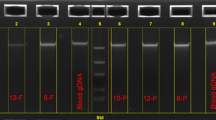
A simple DNA test has been developed for the detection of the AGUFin mutation [7]. This solid-phase minisequencing test is based on amplification of the DNA fragment spanning the mutation using one biotinylated PCR primer, collection of the amplification product on a streptavidin-coated matrix, and primer-guided single nucleotide incorporation at the site of the mutation [7, 8] (fig. 1). The result of the test is obtained as an objective numeric value which unequivocally defines the genotype.

Principle of solid-phase minisequencing: 1 = amplification with one biotinylated primer; 2 = affinity capture on a streptavidin-coated support; 3 = denaturation (removal of the unbiotinylated strand); 4 = annealing of a detection step primer and elongation with one labeled nucleotide; 5 = denaturation; 6 = eluted radioactivity measured in a liquid scintillation counter.
The aim of the present study was to evaluate the applicability of the solid-phase minisequencing technique for large scale screening for AGUFin carriers and to determine the carrier frequency in the representative population samples from northern and southern parts of Finland.
Materials and Methods
A total of 1,160 cord blood samples were collected from newborns at delivery hospitals in southern (Turku University central hospital, 553 samples) and northern (Lappi central hospital, 607 samples) Finland. The blood sampling was combined with that of the routine hypothyroidism screening.
Fifty microliters of the EDTA-blood samples were treated with a rapid cell lysis method [9]. One tenth of the lysed sample was amplified by the PCR and genotyped by the solid-phase minisequencing method using reagents from the AffiGene AGUFin kit (Orion Pharmaceutica, Espoo, Finland). The PCR and minisequencing detection primer sequences and assay conditions have been described previously [7]. The genotype of each sample is defined by the ratio of incorporated 3H-labeled dGTP to that of 3H-dCTP (G/C ratio).
Results
The solid-phase minisequencing method was applied to determine the frequency of the AGUFin allele in a population sample of 1,160 newborns. A sample is typed as homozygous normal when the G/C ratio obtained in the test is below 0.1, as heterozygous (AGUFin carrier) when the ratio ranges from 0.5 to 1 and as homozygous AGU patient when the ratio is above 10 [7]. The clear differences in G/C values from each group allow unequivocal identification of different genotypes. We identified 19 AGUFin heterozygotes and no AGUFin homozygotes. Eight of the heterozygotes were from southern and 11 of them from northern Finland, giving carrier frequencies of 1:69 (95% confidence interval 1:217-1:41) and 1:55 (95% CI 1:133-1:35), respectively (table 1). These frequencies did not differ statistically significantly from each other (0.24; p > 0.50). The range of G/C values in samples from normal individuals was 0.0020–0.093 and in samples from heterozygotes 0.51–1.1.
In preliminary experiments we were not able to successfully apply the present sample treatment by lysis on blood samples collected without an anticoagulant (after removal of the serum for hypothyroidism assay). Therefore a separate EDTA-blood sample had to be collected for the AGUFin assay. Six samples (0.5%) were rejected because of inefficient PCR amplification, most likely due to partial coagulation of the samples.
Discussion
The solid-phase minisequencing technique proved to be a simple and reliable test for detection of carriers of the AGUFin mutation. The technique lends itself to automatization and therefore would easily be applicable to large-scale screening programs. The carrier frequency of AGU in defined populations has so far not been studied. It is well known that the gene causing AGU and those causing the other recessively inherited diseases enriched in the Finnish population [10] are unevenly distributed among the population. Regional differences of the prevalence of AGU, as well as the analysis of the birthplaces of the grandparents of today’s patients have indicated clustering of the AGU gene in the northern and eastern part of the population. A carrier frequency of 1:40 was estimated from the prevalence of AGU cases among the mentally retarded patients in the northern district of the country [11]. A carrier frequency in the same order of magnitude was recently obtained using the minisequencing assay in a quantitative fashion. Aliquots of pooled leukocytes derived from transfusion blood of 1,350 donors were analyzed simultaneously with a standard mixture of normal and AGUFin DNA. The G/C ratios obtained in this analysis corresponded to a carrier frequency of 1:36 [7]. The blood donors were mainly from the capital area of the country.
The present study revealed slightly lower carrier frequencies than expected, 1:55 and 1:69 in the northern and southern population sample, respectively. It is worth noting, however, that the differences between these results and those mentioned above fall within the same range of significance. The small sample sizes may contribute to the results and, as stated above, considerable differences in the regional subpopulations can be expected.
Prospects of AGU carrier screening on a population basis in Finland are favored by several facts. Firstly, the prevalence of the disease is relatively high, with 5–7 new cases identified every year in a population of 5 million, secondly, one mutation is responsible for 98% of the mutant alleles in the population, and finally, a reliable and simple PCR-based DNA test is available for carrier detection at low cost. Total costs of a screening program are of course much higher than costs of the laboratory assay alone. Information and counselling, sample collection and delivery as well as report of the result with subsequent actions in cases of identified carriers must be included in the analysis of the total costs of the program. We have not performed a detailed cost-benefit calculation but it is evident that the total costs of the AGU carrier screening program in the pregnant population in this country would be less than the life-long costs of the AGU cases prevented by the program. Furthermore, AGU is a disease associated with severe, nontreatable mental retardation. Whether screening for AGU carriers in the whole or in a restricted fraction of the population will be put into practice remains to be seen. A tempting vision for the future is to combine the AGUFin carrier test with similar mutation detection of one or more other Finnish genetic diseases in order to increase the efficiency of the program. Recent discoveries by linkage studies of the gene loci for infantile neuronal ceroid lipofuscinosis, INCL [12], myoclonic epilepsy [13] and diastrophic dysplasia [14] provide realistic hope that the gene defects in these diseases will soon be characterized. Specific DNA assays detecting a panel of mutations can then be designed and consequently applied to simultaneous screening for carriers of the Finnish inherited diseases.
References
Autio S: Aspartylglucosaminuria. Analysis of thirty-four patients. J Ment Defic Res Monogr Ser I, 1972.
Pollitt RJ, Jenner FA: Aspartylglycosaminuria. An inborn error of metabolism associated with mental defect. Lancet 1968;ii:253.
Aula P, Autio S, Raivio KO, Rapola J: Aspartylglucosaminuria; in Durand P, O’Brien JS (eds): Genetic Errors of Glycoprotein Metabolism. Berlin, Springer, 1982, pp 122–152.
Autio S, Visakorpi JK, Järvinen H: Aspartylglucosaminuria (AGU). Further aspects on its clinical picture, mode of inheritance and epidemiology based on a series of 57 patients. Ann Clin Res 1973;5:149–155.
Aula P, Raivio KO, Autio S: Enzymatic diagnosis and carrier detection of aspartylglucosaminuria using blood samples. Pediatr Res 1976;10:625–629
Ikonen E, Baumann M, Grön K, Syvänen AC, Enomaa N, Halila R, Aula P, Peltonen L: Aspartylglucosaminuria: cDNA encoding human aspartylglucosaminidase and the missense mutation causing the disease. EMBO J 1991;10:51–58
Syvänen AC, Ikonen E, Manninen T, Bengtström M, Söderlund H, Aula P, Peltonen L: Convenient and quantitative determination of the frequency of a mutant allele using solid-phase minisequencing: Application to aspartylglucosaminuria in Finland. Genomics 1992;12:590–595
Syvänen AC, Aalto-Setälä K, Harju L, Kontula K, Söderlund H: A primer-guided nucleotide incorporation assay in the genotyping of apolipoprotein E. Genomics 1990;8:684–692
Kawasaki ES: Sample preparation from blood, cells and other fluids; in Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds): PCR Protocols. A Guide to Methods and Applications. San Diego, Academic Press, 1990, pp 146–152.
Norio R, Nevanlinna HR, Perheentupa J: Hereditary diseases in Finland; rare flora in rare soil. Ann Clin Res 1973;5:109–141
Aula P, Renlund M, Raivio O, Koskela S-L: Screening of inherited oligosaccharidurias among mentally retarded patients in northern Finland. J Ment Defic Res 1986;30:365–368
Järvelä I, Schleutker J, Haataja L, Santavuori P, Puhakka L, Manninen T, Palotie A, Sandkuijl LA, Renlund M, White R, Aula P, Peltonen L: Infantile neuronal ceroid lipofuscinosis (INCL, CLN1) maps to the short arm of chromosome 1. Genomics 1991;8:170–173
Lehesjoki A-E, Koskiniemi M, Sistonen P, Miao J, Hästbacka J, Norio R, de la Chapelle A: Localization of a gene for progressive myoclonus epilepsy to chromosome 21q22. Proc Natl Acad Sci USA 1991;88:3696–3699
Hästbacka J, Kaitila I, Sistonen P, de la Chapelle A: Diastrophic dysplasia gene maps to the distal long arm of chromosome 5. Proc Natl Acad Sci USA 1990;87:8056–8059
Acknowledgements
We thank Ilona Carlsson for technical assistance. This study was supported by the Academy of Finland.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Hietala, M., Grön, K., Syvänen, AC. et al. Prospects of Carrier Screening of Aspartylglucosaminuria in Finland. Eur J Hum Genet 1, 296–300 (1993). https://doi.org/10.1159/000472427
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1159/000472427
Key Words
This article is cited by
-
Aspartylglycosaminuria: a review
Orphanet Journal of Rare Diseases (2016)
