
Abstract
Genetic imprinting is an important component of X chromosome inactivation, since in marsupials and extraembryonic cell lineages of mice and rats, the paternally derived X chromosome is preferentially inactivated. This imprinting is thought to be mediated via the X inactivation centre. The gene symbolized Xist is a strong candidate for a role in the function of the X inactivation centre and the paper reviews the evidence that Xist shows imprinted behaviour and that differential methyiation is the possible basis of the imprint.
This paper is the text of the speech given by Dr. Mary Lyon after the awarding of the Mauro Baschirotto prize at the meeting of the European Society of Human Genetics in Paris, June 1994 (see page 305).
Similar content being viewed by others


In X inactivation in eutherian mammals typically either of the two X chromosomes is inactivated in different cells of the same animal, leading to the characteristic variegated phenotype seen in females heterozygous for X-linked genes [reviewed in ref. 1–3]. However, in marsupials the picture is different, with the paternally derived X chromosome being preferentially inactivated in all cells [reviewed in ref. 4]. In addition, in mice and rats similar preferential inactivation of the paternally derived X chromosome is seen in two extraembryonic cell lineages, the trophectoderm and primitive endoderm [5]. Thus, imprinting is an important component in X chromosome inactivation. To understand something of the association of the two phenomena one has to consider the mechanism of X inactivation.
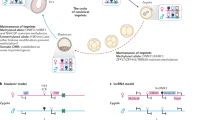
In the very early mouse embryo, both X chromosomes in each cell are active. Inactivation is initiated first in the trophectoderm and primitive endoderm, the two lineages which show imprinting, at the late blastocyst stage. Somewhat later, at the egg cylinder stage, inactivation takes place in the primitive ectoderm, which gives rise to the embryo proper, where the choice of X chromosome is random (fig. 1). Once inactivation has occurred it remains stable throughout all further cell divisions in the lifetime of the animal, except in the female germ cell, where reactivation occurs around the time of meiosis.

Changes in X chromosome activity during the life cycle of the mouse. The diagram shows the correlation of Xist expression with inactivation, and with the presence of imprinting. Me = Methylation of CpG islands of genes on the inactive X chromosome (not methylation of the Xist gene). Reprinted with permission from Lyon [28].
According to present thinking, the mechanism of X inactivation has three parts, the first of which is initiation of inactivation in the early embryo. The second component is spreading of the inactivation to cover the whole, or almost the whole, chromosome and the third is the maintenance of inactivation throughout all subsequent cell divisions. The initiation of X inactivation is thought to occur via the X inactivation centre.
The X Inactivation Centre and the Xist Gene
Evidence about the inactivation centre has come from mouse X-autosome translocations. When autosomal coat colour genes are translocated to the X chromosome, inactivation spreads into the attached autosomal material and causes variegation for these colour genes, but it only spreads into one of the two autosomal segments. In addition, only one of the two segments into which the X is broken shows late replication and dark Kanda staining, and X-linked genes on one segment remain active in all cells. The interpretation is that the segment that lacks an inactivation centre cannot undergo inactivation [reviewed in ref. 6, 7].
In individuals with supernumerary X chromosomes, e.g. XXY, XXXX, a single X chromosome remains active, and all others become inactive. Thus, there is considered to be a counting mechanism, such that one X chromosome remains active per two autosome sets, and this mechanism is thought to act on the X inactivation centre. One centre receives some blocking factor, and the chromosome with the blocked centre remains active. All other centres in the cell (normally one) then initiate inactivation which spreads in a cis-limited manner along the chromosome in both directions. Segments without a centre remain active.
By studying different translocations and deletions it has been possible to map the centre to band XD in mouse and band Xq13 in humans. A human gene was cloned from the relevant region, with the unique property of being expressed from the inactive but not the active X chromosome [8]. The gene was termed XIST (X-inactive-specific transcript). A homologous gene (Xist) with the same properties of expression was also cloned from the region of the centre in the mouse [9, 10]. From the combination of its location and its unique pattern of expression, XIST/Xist is a strong candidate for a role in the X inactivation centre, and the first hypothesis would be that the imprinting seen in X chromosome inactivation is mediated through the Xist locus.
Before considering the imprinting of Xist, it is important to deal with the evidence that it may indeed be part of the inactivation centre. For Xist to have a causal role in X inactivation it must be expressed at appropriate stages in development. Kay et al. [11] studied the expression of Xist in the very early mouse embryo. No expression was seen in 2-cell embryos, but by the 8-cell and morula stages, expression was already present, and continued through the blastocyst and egg cylinder stages. X chromosome inactivation first occurs in the trophectoderm at the late blastocyst stage, at least a day later than the first appearance of Xist expression [12]. By breeding embryos in which the two parentally derived alleles of Xist were distinguishable, Kay et al. [11] also showed that at first only the paternally derived allele (Xp) was expressed. Expression of the maternally derived allele (Xm) appeared later. Thus, Xist is expressed before the initiation of X inactivation, its first expression is imprinted, but random expression occurs later. In all these aspects, the pattern of Xist expression is such that it could have a causal role in X inactivation. However, since expression was seen as early as the 8-cell stage, and even earlier, at the 4-cell stage, in later work, when both X chromosomes are active, expression of Xist is clearly not sufficient for X inactivation. Some other factor must also be involved.
Other authors have studied Xist expression in germ cells. In male germ cells, the single X chromosome becomes inactive early in spermatogenesis. In contrast to the female, in which Xist is expressed in all adult tissues, in the male no Xist expression is seen except in the testis and specifically in the germ cells [13–15]. In the female, McCarrey and Dilworth [15] showed that Xist expression disappeared from germ cells at around the time of reactivation of the X chromosome at meiosis. Thus, as in the embryo, the expression of Xist is appropriate for it to have a causal role in the change of X chromosome activity, but the evidence that it in fact has such a role is not yet complete.
A further situation in which Xist expression changes with X chromosome activity is in embryonic stem (ES) cells. In these cell lines, both X chromosomes are active while the cells are maintained in a totipotent state, but if the cells are allowed to differentiate, X chromosome inactivation occurs. Kay et al. [11] showed that differentiation of such an XX ES cell line was accompanied by Xist expression.
Evidence Concerning Imprinting
Rastan and colleagues [16, 17] have carried out two series of experiments on Xist gene imprinting. In the first, they considered the possible role of DNA methyiation in imprinting and expression of Xist. DNA methyiation has been proposed as a possible mechanism of imprinting, and the inactive X chromosome is known to show differential methyiation of CpG islands, and hence the question of methyiation of the Xist gene in different cell types is highly relevant. Norris et al. [17] found that in adult male mice, where the Xist gene on the single active X chromosome is not expressed, it was fully methylated at sites in the promoter and 5′ region of the body of the gene. Conversely, in the female, both methylated and unmethylated forms of the corresponding sites were present. By breeding females with a distinguishable Xist allele on each X chromosome, Norris et al. [17] showed that the active X chromosome carried a fully methylated Xist gene, whereas the Xist allele on the normal inactive X chromosome was completely unmethylated.
In yolk sac endoderm tissue of normal females, in which the paternal X chromosome is preferentially inactivated, the paternal, expressed, allele of Xist was completely unmethylated and the maternal allele fully methylated.
These results indicated that methyiation was associated with a lack of transcription of Xist. In order to test whether methyiation might also be associated with imprinting of Xist, Norris et al. [17] studied male germ cells. They found that methyiation of Xist was lost at around the time of entry into meiosis and this demethylated state of Xist was retained in mature sperm. Hence, the paternal gamete entering the new zygote was presumably also demethylated, and thus differential methyiation could constitute the parental imprint on the Xist gene.
Further insight into the imprinting of Xist has come from studies of androgenotes, gynogenotes and parthenogenotes. Earlier work on parthenogenotes had shown that the counting mechanism maintaining one X chromosome active per two autosome sets can override the parental origin of chromosomes. Thus, in diploid parthenogenotes with two maternally derived X chromosomes, inactivation of one still occurs, even in extraembryonic tissues, and in XO females with a paternally derived X chromosome this remains active in extraembryonic as well as embryonic tissues. Kay et al. [16] studied the expression of Xist in parthenogenetic and gynogenetic embryos with two maternally derived X chromosomes. Whereas in normal embryos Xist expression first appeared at the 4-cell stage, in parthenogenotes and gynogenotes it was delayed, first appearing at the morula and the blastocyst stages, respectively. In embryos with distinguishable alleles of Xist, cells expressing one or other occurred equally. By contrast, in androgenotes with only Xp chromosomes, and therefore unmethylated Xist alleles, expression of Xist began at the 4-cell stage. Thus, this evidence suggests that the parental imprints on Xist indeed determine its expression in the early embryo, and the imprint appears to be lost by the blastocyst stage.
The androgenetic embryos yielded further interesting results. Whereas all gynogenetic embryos are XX, androgenetic embryos can be chromosomally of three types: XpXp, XpY and YY. The YY embryos would be expected to be eliminated early. The results suggested that all the remaining embryos, both XpXp and XpY, were expressing Xist. Thus, the counting mechanism maintaining one Xist allele inactive was apparently not functional. Thus, as early development proceeds, the parental imprint on Xist is lost at the same time as the counting mechanism develops. A further difference from normal embryos lay in the duration of Xist expression. Whereas normally Xist expression is first seen at the 4-cell stage and then persists, in the androgenetic embryos down-regulation began at the late 8-cell stage and expression had disappeared by the blastocyst stage. The authors conclude that in normal embryos some other maternally inherited factor maintains the expression of Xist, and that this is missing in androgenetic embryos.
Methylation and X Inactivation
If differential methylation forms the imprint on Xist, it is interesting to consider the role of methylation in the X inactivation process. The suggestion was put forward by various authors that methylation might form the mechanism of spreading of inactivation along the chromosome. It is known that CpG islands in the 5′ region of genes on the inactive X chromosome are heavily methylated, in contrast to their unmethylated state in the genome generally [3, 18]. However, this methylation is not seen in all types of X inactivation, being absent in marsupials [4], in mouse germ cells [19] and possibly also in extraembryonic lineages (fig. 1) [19, 20]. In addition, it appears after the first initiation of inactivation [19, 21]. It is therefore thought to be part of the mechanism stabilizing inactivation, rather than the spreading mechanism.
Function of the Xist Gene
If differential methylation is not the spreading mechanism, other possibilities include some change in the structure of the chromatin fibre or in binding of proteins to chromatin. The sequence of the Xist gene has been determined with a view to understanding its role in spreading inactivation. The structure of the gene is well conserved among mammals [22, 23]. In both human and mouse, the gene has no substantial open reading frame, and the gene product is retained in the nucleus. Thus, the gene may act through RNA, possibly by preventing the binding of proteins or by altering the chromatin configuration in the region.
Possible Evolutionary Association of Imprinting and X Inactivation
A further question concerns the association of imprinting and X inactivation in evolution. An early suggestion was that in primitive mammals X inactivation in somatic cells of females had evolved from an earlier inactivation of the X chromosome in male germ cells [24, 25]. Inactivation of heteromorphic sex chromosomes during meiosis is seen in various animal groups in which the male is the heterogametic sex, and clearly has some selective advantage, possibly in protecting unsaturated pairing sites. The suggestion was that inactivation of the paternal X chromosome in spermatogenesis was carried over into the embryo and provided a starting point for somatic X inactivation. The discovery of the possible demethylation imprint on the Xist gene in sperm, leading to the differential expression of the paternal allele of Xist in the early embryo, provides evidence in favour of this suggestion. In the very early embryo, the mechanism for counting X chromosomes is apparently not functional. In a normal animal, imprinting would be enough to give the observed single X chromosome activity, without the need for a counting mechanism. The single Xm in XmY males would be active and the Xp of XmXp females inactive. In eutherians, from observation of X chromosome aneuploids and polyploids, it is known that there is a counting mechanism. However, in marsupials it is not really clear if a counting mechanism is present, since far fewer sex chromosome aneuploids are available for study. It may be that the primitive form of X inactivation was the imprinted type and that the counting mechanism only arose with the switch to random inactivation. The imprinted type of X inactivation is less stable than the random type. In marsupials, reactivation of the paternal allele of X-linked genes may occur in cultured cells or in some tissues in vivo [4]. It is possible that there is some feature of imprinted X inactivation that makes instability inherent and that the selective advantage that led to the evolution of random inactivation lay in its providing more stable inactivation and hence better dosage compensation.
Thus, the hypothetical picture would be that in evolution, the inactivation of the primitive X chromosome first took place in male germ cells, as the X and Y began to differentiate. This resulted in differential methyiation of the Xist locus, which when carried over into the very early embryo led to differential expression of the X chromosome. In present mammals, the function of X inactivation seems clearly to be dosage compensation. Activity of additional X chromosome material in aneuploids and translocation heterozygotes is clearly harmful [26, 27]. However, it is possible that dosage compensation was not the original function of X inactivation. In mouse embryos with activity of supernumerary X chromosomes, the extraembryonic tissues are poorly developed, as in parthenogenotes [27]. It is possible that in evolution the inactivation of the paternal X chromosome favoured the development of the extraembryonic tissues and thus was involved in the evolution of viviparity. It may be that only later was the function of dosage compensation added, and that this was still later associated with the development of a random, more stable type of X inactivation in eutherians.
Thus, it is now possible to make many speculations as to the mechanism and evolution of X inactivation. However, this all turns on the Xist gene, and its role in the X inactivation centre. The circumstantial evidence in its favour is now strong and we must hope that firm evidence will become available soon.
References
Lyon MF: Some milestones in the history of X-chromosome inactivation. Annu Rev Genet 1992;26:15–27
Lyon MF: X-chromosome inactivation; in Wachtel SS (ed): Molecular Genetics of Sex Determination. San Diego, Academic Press, 1994, pp 123–142.
Gartier SM, Dyer KA, Goldman MA: Mammalian X-chromosome inactivation; in Friedman T (ed): Molecular Genetic Medicine. New York, Academic Press, 1992, vol 2, pp 121–160.
Cooper DW, Johnston PG, Watson JM, Graves JAM: X-inactivation in marsupials and monotremes. Semin Dev Biol 1993;4:117–128
Takagi N, Sasaki M: Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse. Nature 1975;256:640–642
Lyon MF: The William Allan memorial award address: X-chromosome inactivation and the location and expression of X-linked genes. Am J Hum Genet 1988;42:8–16
Rastan S, Brown SDM: The search for the mouse X-chromosome inactivation centre. Genet Res 1990;56:99–106
Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF: A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991;349:38–44
Borsani G, Tonlorenzi R, Simmler MC, Dandolo L, Arnaud D, Capra V, Grompe M, Pizzuti A, Muzny D, Lawrence C, Willard HF, Avner P, Ballabio A: Characterization of a murine gene expressed from the inactive X chromosome. Nature 1991;351:325–329
Brockdorff N, Ashworth A, Kay GF, Cooper P, Smith S, McCabe VM, Norris DP, Penny GD, Patel D, Rastan S: Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 1991;351:329–331
Kay GF, Penny GD, Patel D, Ashworth A, Brockdorff N, Rastan S: Expression of Xist during mouse development suggests a role in the initiation of X chromosome inactivation. Cell 1993;72:171–182
Takagi N, Sugawara O, Sasaki M: Regional and temporal changes in the pattern of X-chromosome replication during the early post-implantation development of the female mouse. Chromosoma 1982;85:275–286
Richler C, Soreq H, Wahrman J: X inactivation in mammalian testis is correlated with inactive X-specific transcription. Nat Genet 1992;2:192–195
Salido EC, Yen PH, Mohandas TK, Shapiro LJ: Expression of the X-inactivation-associated gene Xist during spermatogenesis. Nat Genet 1992;2:196–199
McCarrey JR, Dilworth DD: Expression of Xist in mouse germ cells correlates with X-chromosome inactivation. Nat Genet 1992;2:200–203
Kay GF, Barton SC, Surani MA, Rastan S: Imprinting and X chromosome counting mechanisms determine Xist expression in early mouse development. Cell 1994;77:639–650
Norris DP, Patel D, Kay GF, Penny GD, Brockdorff N, Sheardown SA, Rastan S: Evidence that random and imprinted Xist expression is controlled by preemptive methylation. Cell 1994;77:41–51
Riggs AD, Pfeifer GP: X-chromosome inactivation and cell memory. Trends Genet 1992;8:169–174
Grant M, Zuccotti M, Monk M: Methylation of CpG sites of two X-linked genes coincides with X-inactivation in the female mouse embryo but not in the germ line. Nat Genet 1992;2:161–166
Kratzer PG, Chapman VM, Lambert H, Evans RE, Liskay RM: Differences in the DNA of the inactive X chromosomes of fetal and extraembryonic tissues of mice. Cell 1983;33:37–42
Lock LF, Takagi N, Martin GR: Methylation of the Hprt gene on the inactive X occurs after chromosome inactivation. Cell 1987;48:39–46
Brockdorff N, Ashworth A, Kay GF, McCabe VM, Norris DP, Cooper PJ, Swift S, Rastan S: The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992;71:515–526
Brown CJ, Hendrich BD, Rupert JL, Lafreniere RG, Xing Y, Lawrence J, Willard HF: The human XIST gent: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992;71:527–542
Cooper DW: A directed genetic change model for X-chromosome inactivation in eutherian mammals. Nature 1971;231:292–294
Lifschytz E, Lindsley DL: The role of X-chromosome inactivation during spermatogenesis. Proc Natl Acad Sci USA 1972;69:182–186
Shao C, Takagi N: An extra maternally derived X chromosome is deleterious to early mouse development. Development 1990;110:969–975
Tada T, Takagi N, Adler ID: Parental imprinting on the mouse X chromosome: Effects on the early development of XO, XXY and XXX embryos. Genet Res 1993;62:139–148
Lyon MF: Epigenetic inheritance in mammals. Trends Genet 1993;9:123–128
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Lyon, M.F. The X Inactivation Centre and X Chromosome Imprinting. Eur J Hum Genet 2, 255–261 (1994). https://doi.org/10.1159/000472369
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1159/000472369
