
Abstract
Bovine viral diarrhea virus (BVDV) is considered to be the most common agent of severe diarrhea in cattle worldwide, causing fever, diarrhea, ulcers, and abortion. Bovine herpesvirus 1 (BoHV-1) is also a major bovine respiratory disease agent that spreads worldwide and causes extensive damage to the livestock industry. Recombinase polymerase amplification (RPA) is a novel nucleic acid amplification method with the advantages of high efficiency, rapidity and sensitivity, which has been widely used in the diagnosis of infectious diseases. A dual RPA assay was developed for the simultaneous detection of BVDV and BoHV-1. The assay was completed at a constant temperature of 37 °C for 30 min. It was highly sensitive and had no cross-reactivity with other common bovine viruses. The detection rate of BVDV RPA in clinical samples (36.67%) was higher than that of PCR (33.33%), the detection rate of BoHV-1 RPA and PCR were equal. Therefore, the established dual RPA assay for BVDV and BoHV-1 could be a potential candidate for use as an immediate diagnostic.
Similar content being viewed by others


Introduction
Bovine viral diarrhoea viruses (BVDV), with positive, non-segmented, single stranded RNA genome, comprise a heterogeneous group of viruses which belong to the genus Pestivirus, the Flaviviridae family, and cause bovine viral diarrhea-mucosal disease (BVD-MD)1. BVD-MD is an acute, febrile, contact infectious disease, clinically manifests as acute diarrhea, respiratory disease, immunosuppression, and reproductive disorders2. BVDV has a wide host range, its infection has been detected not only in domesticated ruminants, but also in wild ruminants and wild boars3. BVDV is one of the major pathogens leading to the decline in cattle production performance. It is a serious threat to the development of animal husbandry in our country and even globally, resulting in severe economic losses. Bovine herpesvirus 1 (BoHV-1) is a member of the Herpesviridae family and genus Varicellovirus4. BoHV-1 is responsible for causing different syndromes, such as infectious bovine rhinotracheitis (IBR) and infectious pustular vulvo-vaginitis (IPV) in cows and balanoposthitis (IBP) in bulls5. BoHV-1 has been widely prevalent throughout the world with varying rates of positivity since it was first reported in 1953 in dairy cattle feedlots in Los Angeles, California6. To date, some countries have been able to eradicate IBR, such as Scandinavian countries (Denmark, Finland, Norway, and Sweden), as well as Austria, Switzerland, and some Italian regions7. Some studies have shown that BVDV and BoHV-1 usually coexist in diseased cattle. When cattle are infected with one virus, the immune system becomes weak, making it easier for other pathogens to enter the body, which is the main reason why these diseases are so difficult to eradicate8.
So far, detection methods for the diagnosis of these two viruses include virus isolation9, PCR techniques10, serological neutralization tests11, and enzyme-linked immunosorbent assays12. Virus isolation relies on live viruses to determine positive results, is expensive and time consuming13. The molecular biology assays detecting viral genomic DNA are either time-consuming or expensive or require sophisticated laboratory setup and skilled personnel14. The serologic methods depend on high quality antisera which are severely affected by improper sampling and autolysis, and are likewise more expensive15. These methods cannot accomplish rapid detection of clinical samples. Therefore, there is an urgent need to establish a rapid and accurate diagnostic method to provide a new generation of detection technology and means for the detection and control of bovine diseases.
Recombinase polymerase amplification (RPA) is a nucleic acid level detection technique different from PCR, which mainly involves three enzymes, namely, binding single-stranded nucleic acid recombinase (T32 UvsX), single-stranded DNA-binding protein (SSB), and strand-replacing DNA polymerase (Bsu polymerase)16. The RPA assay requires low temperature, short reaction time, simple operation, no special equipment and high stability17. Since its initial development, the RPA assay has been widely used for the detection of human, animal and plant pathogens such as Mycobacterium tuberculosis18, feline herpesviruses19, African swine fever virus20, and Yersinia coli21. The main commonly nucleic acid end-product assays are agarose gel electrophoresis(AGE)16, real-time fluorescence22, chemical color development23, electrochemistry24 and lateral flow test strips (LFD)25. Some studies have combined RPA with LFD for the detection of BoHV-1, but the LDF-RPA assay is relatively costly and not suitable for resource-limited areas26. Isothermal-based diagnostic techniques are single detection systems because they cannot detect multiple viruses at the same time and increase the output time of results in case of mixed infections. Nevertheless, AGE is the most commonly used method for nucleic acid end-product assay due to its lower cost in summer of equivalent conditions and simultaneous detection of multiple pathogens27. Multiple detection systems have been developed to detect multiple targets by detecting them in a single-tube reaction. Studies have reported the development of dual RPAs using animals and humans to detect pathogens including bacterial tick-borne diseases28,Chlamydia trachomatis (CT) and Neisseria gonorrhea (NG) in humans29, and major bacterial pathogens of bovine respiratory diseases30. Although RPA assay of BoHV-1 has been studied, BVDV and BoHV-1 usually present as mixed infections31. Therefore, for the first time, we developed a simple and efficient RPA assay for the simultaneous detection of BVDV and BoHV-1 for the first time, which provides technical support for the diagnosis of bovine diseases and epidemiologic risk assessment.
Results
Construction and characterization of the pcDNA 3.1-BVDV and pcDNA 3.1- BoHV-1 recombinant plasmids
The BVDV 5'UTR was constructed into the pcDNA 3.1 by Bam HI and Hind III restriction sites to obtain the pcDNA 3.1-BVDV recombinant plasmid. The BoHV-1 gB gene sequence was constructed into pcDNA 3.1 plasmid by Hind III and Xhol restriction sites to obtain pcDNA 3.1-BoHV-1 recombinant plasmid. The map of the plasmids is shown in Fig. 1. The results obtained from AGE (1%) of pcDNA 3.1-BVDV and pcDNA 3.1-BoHV-1 recombinant plasmids after digestion are shown in Fig. 2. The positions of the target bands were as expected. The pcDNA 3.1-BVDV recombinant plasmid showed a 365 bp band after double digestion, and the pcDNA 3.1- BoHV-1 recombinant plasmid showed a 493 bp band after double digestion. The sequencing results also matched the sequence of the target gene.

The map of pcDNA 3.1-BVDV and pcDNA 3.1-BoHV-1 recombinant plasmids. (A) pcDNA 3.1-BVDV recombinant plasmid mapping. (B) pcDNA 3.1- BoHV-1 recombinant plasmid mapping.

Recombinant plasmid digest identification. (A) pcDNA 3.1-BVDV digest identification, Lane M. 1 kb plus DNA Marker; Lane 1. Hind III single digest; Lane 2. Hind III-Xhol double digest. (B) pcDNA 3.1-BoHV-1 digest identification Lane M. 1 kb DNA marker; Lane 1. Hind III single digest; Lane 2. Hind III-Bam HI double digest. Insert is highlighted by a red box.
Establishment and optimization of reaction conditions for BVDV and BoHV-1 dual RPA assay
Candidate primers for BVDV, BoHV-1 RPA assay was selected by Twist Amp® Basic reaction and initially analyzed on 2% agarose gel. The BVDV-RPA agarose gel results indicated that primer sets 5'UTR-1F/R and 5'UTR-2F/R produced specific amplification efficiency for RPA assay, both amplifying a 208 bp fragment. The primer set 5'UTR-3F/R amplified a 206 bp fragment, but with more non-specific bands and primer dimerization (Fig. 3A). In addition, the 5'UTR-1F/R primer set amplified the brightes and better amplification of the target band under the same conditions. Therefore, it was subsequently used in the BVDV-RPA assay. The agarose gel results of BoHV-1 RPA assay presented in Fig. 3B. Primer groups gB-1F/R and gB-2F/R produced specific amplification efficiency in the RPA assay, and the expected sizes of the products was 141 or 135 bp. Primer set gB-3F/R did not amplify the target band (139 bp) and showed a very distinct primer dimer that failed to meet the experimental requirements. Under the same conditions, gB-1F/R amplified the brightest and better amplification effect. Therefore, the gB-1F/R primer set was selected for subsequent use in the BoHV-1 RPA assay. Meanwhile, to verify the availability of the designed primers in BVDV and BoHV-1 dual RPA assay, the pcDNA 3.1-BVDV, pcDNA 3.1- BoHV-1 recombinant plasmid premixes were amplified by 5'UTR-1F/R, gB-1F/R, respectively. As a result, the expected 5'UTR (208 bp) and gB (141 bp) gene amplified fragments were successfully observed after using dual gene amplification primers (Fig. 3C).

Primer screening for the dual RPA assay. (A) Primer screening for BVDV-RPA assay, Lane M. 50 bp DNA Marker; Lane 1. 5'UTR-1 primers; Lane 2. 5'UTR-2 primers; Lane 3. 5'UTR-3 primers; Lane 4. Negative control. (B) Primer screening for BoHV-1 RPA assay, Lane M. 50 bp DNA Marker; Lane 1. gB-1 primers; Lane 2. gB-2 primers; Lane 3. gB-3 primers; Lane 4. Negative control. (C) BVDV and BoHV-1 dual RPA primer screen, Lane M. 50 bp DNA Marker; Lane 1. 5'UTR-1 primers; Lane 2. gB-2 primers; Lane 3. 5'UTR-1 primers and gB-1 primers premixes.
The optimal reaction temperature for BVDV and BoHV-1 dual RPA assay were determined by testing different temperatures (30 °C, 32 °C, 35 °C, 37 °C, 39 °C, 42 °C), and reaction times (10 min, 15 min, 20 min, 25 min, 30 min, 40 min). As shown in Fig. 4, the target fragment gradually brightened with increasing reaction temperature, with the strongest color change at 37 °C. After that, the brightness of the target fragment decreased with increasing temperature, which may be caused by the inactivation of enzymes in the system due to high temperature, or high temperature makes the primers anneal less efficiently. Histogram analysis of the gel intensity by Image J analysis also showed that the BVDV and BoHV-1 RPA amplification was most efficient at 37 °C. Therefore, 37 °C was determined as the optimal reaction temperature. In addition, target fragment brightness was greatest at a reaction time of 30 min, and 30 min was the optimal reaction time (Fig. 5). Blank control tubes provided negative results under each condition. Finally, the optimal condition of 30 min reaction at 37 °C was selected for subsequent testing.

Reaction optimization of double RPA temperature conditions. (A) Double RPA temperature optimized gel electrophoresis. Lane M.DNA 50bp marker; Lane NC. Negative control. (B) Dual RPA temperature optimized gel intensity quantification.

Reaction optimization of different time period of double RPA. (A) Dual RPA time-optimized gel electrophoresis. Lane M.DNA 50bp marker; Lane NC. Negative control. (B) Quantification of dual RPA time-optimized gel intensity.
Different temperatures (30 °C, 32 °C, 35 °C, 37 °C, 39 °C, 42 °C) were set to determine the optimal reaction temperature for BVDV and BoHV-1 RPA assay. The negative control was incubated at 4 °C. As shown in Fig. 4, the temperature had a strong effect on the BVDV RPA assay, with the target bands becoming brighter as the temperature increased, and then darker after being brightest at 37 °C. The histogram of the gel intensity analyzed by Image J analysis also presented the same trend. Although the temperature has a small effect on the BoHV-1 RPA assay, the same conclusions can still be drawn as for the BVDV RPA assay. The brightness of the target fragment decreases with increasing temperature, which may be caused by the inactivation of enzymes in the system due to high temperature, or high temperature makes the primers anneal less efficiently. Therefore, 37 °C was determined as the optimal reaction temperature for BVDV and BoHV-1 RPA assays. The same method was applied with different reaction times (10 min, 15 min, 20 min, 25 min, 30 min, 40 min) to determine the optimal reaction time for the BVDV and BoHV-1 dual RPA assay. The negative control reaction time was 0 min. The BVDV RPA assay had almost no target bands and weak gel intensity after 10 min of amplification. Over time, the target fragment brightness was maximized at a reaction time of 30 min. The BoHV-1 RPA assay showed less variation over time, and quantitative analysis of gel strength revealed that gel strength was greatest at 30 min relative to other times (Fig. 5). Therefore, we chose the optimal condition of 30 min at 37 °C for the subsequent BVDV and BoHV-1 dual RPA assay.
Sensitivity and specificity evaluation of the dual RPA assay
The quantified pcDNA 3.1-BVDV and pcDNA 3.1-BoHV-1 recombinant plasmids were serially diluted from 1 × 1010 copies/μL to 1 × 100 copies/μL. The sensitivity test for BVDV and BoHV-1 dual RPA assay was performed by the electrophoretic detection technique. Nuclease-free water was used as a negative control. As shown in Fig. 6, the minimum detection limit for BVDV RPA-AGE was 1 × 101 copies/μL, and that for BoHV-1 RPA-AGE was 1 × 104 copies/μL.

Sensitivity evaluation of dual RPA assay. (A) Gel electrophoresis diagram of double RPA sensitivity experiment. Lane M.DNA 50bp marker; Lane NC. Negative control. (B) Quantification of gel strength for double RPA sensitivity assay.
The specificity of the dual RPA assay for BVDV and BoHV-1 was verified by RPA assays for six bovine respiratory and diarrhea-associated viruses (BVDV, BoHV-1, BCoV, BRV, BNoV, and BAstV). The results demonstrated that BVDV and BoHV-1 could be specifically detected using the established dual RPA. The distinctive bands of 5'UTR or gB were clearly visible in the gel. No bands were amplified for other pathogens, such as BCoV, BRV, BNoV, BAstV, and nuclease-free water (Fig. 7). In summary, the established RPA assay can be used to specifically detect BVDV and BoHV-1 in single infection or co-infection.

Specificity evaluation of the BoHV-1 RPA assay. (A) Specificity evaluation of the dual RPA assay. Lane M. DNA 50bp marker; Lane NC. Negative control. (B) Quantification of gel strength for the dual RPA specificity experiment.
Dual RPA assay for BVDV and BoHV-1 in clinical samples
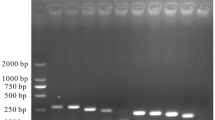
To evaluate the practical clinical application of the dual RPA assay for BVDV and BoHV-1, we performed the RPA assay on nasal swabs, anal swabs, and serum samples from cattle with significant respiratory and diarrheal disease. At the same time, the samples were tested using PCR to verify the RPA assay results. The results of RPA assay of clinical samples are shown in Fig. 8 and Table 1. The total positive rate of BVDV RPA assay of clinical samples was 36.67% (11/30). The positive rates of RPA in bovine nasal swabs, anal swabs and blood serum samples were 20%, 40% and 50%, and the positive rates of PCR were 20%, 30% and 50%. The positive rates of RPA and PCR assay in BoHV-1 clinical samples were the same as those of PCR, which were 33.33% (10/30). The RPA positivity of bovine nasal swabs, anal swabs and blood serum samples were 40%, 30% and 30%. Quantitative analysis of agarose gels revealed that the gel strength of the RPA assay was generally higher than that of PCR assay. Therefore, the established dual RPA method can be used as a candidate assay for the detection of BVDV and BoHV-1 in clinical samples.

Dual RPA clinical sample detection. (A) Agarose gel plot of bovine clinical samples for dual RPA assay of BVDV and BoHV-1. The upper band is BVDV (208bp) and the lower band is BoHV-1 (141bp). (B) Agarose gel plot of BVDV and BoHV-1 PCR assays in bovine clinical samples. The upper band is BoHV-1 (481bp) and the lower band is BoHV-1 (354bp). (C) Gel strength quantification by BVDV and BoHV-1 dual RPA assays. (D) Gel strength quantification for BVDV and BoHV-1 PCR assays. Lane 1–10. Bovine clinical samples; Lane 11. Negative control; Lane 12. Positive control.
Discussion
BVDV and BoHV-1 are important pathogens in cattle with a global distribution that can cause significant economic losses32. BVDV isolates are further stratified into 3 species, BVDV-1 (1a–1 w, 23), BVDV-2 (2a–2d), and HoBi-like viruses (a–d)1. BVDV-1 has the highest occurrence in cattle population in China33. Therefore, rapid diagnosis of BVDV-1 epidemic strains has important significance in epidemiological studies. The 5′UTR of BVDV is most often targeted for molecular diagnosis technology and genotyping since it is highly conserved in the pestivirus genome34. According to its antigenic and genomic characteristics, BoHV-1 is further subdivided into two distinct yet closely related subtypes: 1 (BoHV-1.1) and 2 (BoHV-1.2). Such subtypes may also be associated with distinct manifestations of the disease in cattle35. The gB gene is highly conserved among members of the Herpesviridae family and is frequently used as the target gene to detect BoHV-136. During the development of the RPA assay, we chose the gB gene to design the primers.
Although vaccines against both viruses are available, such as the traditional modified live vaccine (MLV), inactivated vaccine (KV), DIVA (differentiate infected from vaccinated animals) vaccine and subunit vaccine37. However, the prevention and control of bovine diseases still require the establishment of rapid and reliable nucleic acid detection methods. RPA, an emerging isothermal nucleic acid amplification technique, has been applied to detect pathogens in a variety of samples, e.g., blood38, food39, and feces40. In our study, we established a dual RPA assay based on the BVDV 5'UTR and BoHV-1 gB genes and validated it in bovine nasal swabs, anal swabs, and blood samples. In contrast, conventional PCR reactions require agarose gel electrophoresis and fluorescent quantitative PCR methods require thermal cyclers. In terms of reaction time, the RPA method (< 45 min) is less time-consuming than the PCR method41. In terms of experimental cost, compared to the cost of conventional PCR and real-time fluorescent quantitative PCR, although the RPA platform is more costly.
In addition to rapidity, specificity, and cost, we also tested the sensitivity of the RPA assay. The minimum detection threshold for BVDV-AGE was 1 × 101 copies/μL, and that for BoHV-1-AGE was 1 × 104 copies/μL. We established a dual PRA assay with a lower detection threshold for BVDV than RT-LAMP (4.67 × 100) but higher than RT-PCR (4.67 × 103), and a lower detection limit for BoHV-1 than qPCR (4.45 × 102) but higher than LAMP (2 × 104)42,43. The overall positivity rate for the BVDV RPA assay was higher in the clinical trials (36.67%) than the rate of the PCR assay (33.33%). One anal swab sample was positive by RPA assay and negative by PCR assay. Not only that, all other RPA gel electrophoresis intensities that tested positive were significantly higher than those of the PCR assay. It was found that the positive rate of serum samples was higher than that with anal swabs. The higher positivity rate of anal swabs than nasal swabs indicates that BVDV is present in the intestine as the major bovine diarrhea virus. The high positivity rate of blood serum samples could only be attributed to the presence of viral nucleic acid in the blood of cattle, not live virus. The positivity rate of bovine nasal swabs for BoHV-1 RPA and PCR were higher than that of anal swabs and serum samples, which also indicated that BoHV-1 mainly infected the respiratory tract of cattle.
However, the RPA assay still has some limitations. First, RPA assay may have false positives, and reaction tubes should be opened and closed carefully and gloves should be changed frequently. Second, the technique is expensive and not very popular. To reduce the cost, premixing of the RPA reaction system can be done in advance to reduce the total volume to 12.5 μL (1/4 of the original volume). In addition, the RPA assay also avoids nucleic acid extraction, thus allowing the user to easily perform multiple diagnostic assays44, which can be used to further reduce the cost of RPA assay. The success of multiplex RPA for simultaneous diagnosis of BVDV and BoHV-1 not only maintains the accuracy and sensitivity of the RPA assay, but also reduces the number of reagents and procedures required.
Conclusion
In this study, an RPA assay was established for rapid, sensitive, and simultaneous detection of BVDV and BoHV-1 without the need for expensive laboratory equipment. The established RPA assay offers several advantages over other current detection techniques. The RPA method is time-saving and can be completed in less than 30 min. When the template plasmid was diluted to 1 × 101 copies/μL using the RPA method, it demonstrated high sensitivity. The RPA technique does not require thermal cycling and is suitable for non-instrumented nucleic acid amplification platforms. In conclusion, this is a rapid and effective method for the simultaneous detection of BVDV and BoHV-1, especially for resource-limited laboratories. To the best of our knowledge, this study is the first report on simultaneous detection of BVDV and BoHV-1 using RPA.
Methods
The appropriate ethics declarations
The experimental protocol for the collection of bovine clinical samples was strictly in accordance with the Procedures and Guidelines for Animal Ethics in the People's Republic of China. The animal experimental protocol was approved by the Science and Technology Ethics Committee of Ningxia University (Code: NXU-2024-003).
All methods are reported in accordance with ARRIVE guidelines.
Viruses and clinical samples
The BVDV 5'UTR gene sequence information was based on the BVDV-1 strain (MA/101/05, GenBank ID: MW054940.1) and the BoHV-1 gB gene sequence information was based on the BoHV-1 strain (BRV/2018/SMU6352, GenBank ID: MK654723.1). BVDV, BoHV-1, Bovine coronavirus (BCoV/CH/NX-GY-1/2022, GenBank ID: OQ513841.1), bovine rotavirus (BRV/CH/NX-1/2022, GenBank ID: OQ513855.1), and bovine norovirus (BNoV/CH/NX-GY-5/2022, GenBank ID: OQ430676.1), and bovine astrovirus (BAstV/CH/NX- 1/2022) used for specificity evaluation in this study were kept by the laboratory.
The clinical samples were 30 samples of cattle with respiratory disease or diarrhea syndrome collected from different farms in the Ningxia region (July 2022), including 10 nasal swabs, 10 anal swabs, and 10 serum samples. The cattle sampled were approximately 24-week-old bulls of the Simmental breed and had no history of BVDV and BoHV-1 vaccination. All samples were stored at -80 °C before being used for further testing and analysis.
Construction of standard plasmids
The BVDV 5'UTR gene sequence and the BoHV-1 gB gene sequence were synthesized artificially due to the limitation of the pathogen source. The whole genome sequences of BVDV and BoHV-1 were collected in GenBank, and the sequences were compared and analyzed by Mega software. The intraspecies-conserved and interspecies-specific 5'UTR (GenBank ID: MW054940.1) and gB gene (GenBank ID: MK654723.1) were selected as target genes for the RPA assay. The target genes were constructed into pcDNA-3.1 by Hind III, Xhol, and Bam HI enzymes (NEB, No. R0104/ R0146/ R0136), and transformed into E. coli recipient cells Top10, obtain pcDNA 3.1-BVDV, pcDNA 3.1-BoHV-1 recombinant plasmids.
Plasmid extraction was performed according to the Axygen Plasmid Extraction Kit (item no. AP-MN-P), and the concentration measured by Nanodrop ND-8000 spectrophotometer (Thermo Scientific, Dreieich, Germany). The DNA copy number was calculated according to the following formula: DNA copy number = (M × 6.02 × 1023 × 10–9)/ (n × 660), where M is the amount of DNA in nanograms, and n is length of the plasmid in bp. DNA standards were stored at −20 °C until further experiments.
Primer design
The RPA primers were manually designed based on the conserved regions of the BVDV 5'UTR gene, BoHV-1 gB gene, with reference to the instruction manual provided by the RPA reaction manufacturer Twist Dx (Cambridge, UK). Meanwhile, common PCR primers were designed for the constructed plasmid sequences (Table 2). All primers were synthesized by Sangon Biotech (Shanghai, China) and screened by observing their performance on 2% agarose gels.
Establishment of a dual RPA assay for BVDV and BoHV-1
The RPA assay was performed using the extracted recombinant plasmids pcDNA 3.1-BVDV and pcDNA 3.1-BoHV-1 as templates according to the instructions of the Twist Amp® Basic RPA kit (Twist Dx, item no. 10270-106). The following RPA reaction system was configured according to the recommendations of the kit: 2 μL of template, 11.2 μL of sterile deionized water, 29.5 μL of RPA reaction buffer, 2.4 μL each of the F and R primers at 10 pM, and 2.5 μL of magnesium acetate solution, which was mixed homogeneously and then fully solubilized in 4 mg of RPA basic lyophilisate. The RPA reaction was programmed to incubate at 39 °C for 20 min in a thermocycler (Bio Metra GmbH, 844-070-882). The amplification products were subjected to 2% agarose gel electrophoresis and the results were visualized and photographed in a UV gel imaging system.
The main parameters of the RPA assay were amplification temperature and reaction time. To optimize the BVDV, BoHV-1 dual RPA assay, experiments were performed at different reaction temperatures (30 °C, 32 °C, 35 °C, 37 °C, 39 °C, 42 °C) and reaction times (10 min, 15 min, 20 min, 25 min, 30 min, 40 min) according to the manufacturer's recommended protocol. The optimal reaction was determined based on the specific bands in the agarose gel. To quantify the gel imaging results, the target bands (grayscale images of line regions and rectangular selections) were analyzed using ImageJ software. Intensity data were analyzed with Graphpad Prism.
Sensitivity, and specificity evaluation of BVDV and BoHV-1 dual RPA assay
The specificity of the dual RPA assays for BVDV and BoHV-1 was evaluated in bovine pathogens with similar clinical signs. BVDV, BoHV-1, BCoV, BRV, BNoV, and BAstV were detected according to established RPA assays. Nuclease-free water was used as a non-template control in the assay.
To investigate the sensitivity of the dual RPA assay for BVDV and BoHV-1, the quantified pcDNA 3.1-BVDV and pcDNA 3.1-BoHV-1 plasmids were diluted in 11 gradients, i.e., 1 × 1010 copies/μL to 1 × 100 copies/μL. The negative control template was still nucleic acid-free water.
Evaluation of the dual RPA assay using field samples
To effectively examine the effect of the dual RPA assay for BVDV and BoHV-1 established in this study on field samples, we tested 30 samples (10 nasal swabs, 10 anal swabs, and 10 sera) of bovine respiratory syndrome and diarrhea syndrome with obvious clinical symptoms in Ningxia (collected in July 2022) by RPA. The nuclease-free water was used as a negative control, the synthesized recombinant plasmids (pcDNA 3.1-BVDV and pcDNA 3.1-BoHV-1) were used as a positive control. In addition, a conventional PCR assay was performed on the same samples to compare the results with the RPA assay.
The BVDV conventional PCR primers are as follows. 5'UTR-F: TCTCGACCGGGGACATTATCT; 5'UTR-R: CATTCTGCAACGCGAAGGTG. The amplification size is 354 bp. The BoHV-1 conventional PCR primers are as follows. gB-F: GTACGACTCGTTCGCGCTCT; gB-R: CAAGTACGTCTCCAGGCTGCC. The amplification size is 481 bp.
Swab samples were generally resuspended in virus preservation solution. They are shaken for 10 min, centrifuged at 12,000 rpm for 10 min to remove solid precipitates, and filtered aseptically through a 0.22 μm membrane. Blood samples were centrifuged at 3000 rpm for 10 min after an overnight incubation at 4 °C, and the upper serum was aspirated for subsequent tests. The viral genomic DNA/RNA was extracted by the TianGen Viral Genomic DNA/RNA Extraction Kit (No. DP315) according to the manufacturer's instructions. The RNA was reverse transcribed into cDNA by the Prime Script™ II 1st Strand cDNA Synthesis Kit (No. 6210A) from TAKARA. The reverse transcribed cDNA and extracted DNA were subjected to RPA assay or PCR assay. The amplified products were detected by 2% agarose gel electrophoresis and quantitatively analyzed by ImageJ software.
Statistical analysis
To visually determine the specificity, sensitivity, and evaluation of dual RPA in clinical samples, we quantified the gel imaging results and analyzed the target strips (grayscale images of line regions and rectangular selections) using ImageJ software. Intensity data were analyzed with Graphpad Prism. In addition, we cropped the gel imaging results and the original full gel map imaging is shown in the Supplementary Information.
Data availability
The dataset analyzed during the current study is available from the corresponding author on reasonable request. The nucleotide sequences under the relevant accession numbers (MW054940.1, MK654723.1) analyzed during the current study are available in the GenBank repository, https://www.ncbi.nlm.nih.gov/nucleotide/.
References
Ridpath, J. F. Encyclopedia of Virology. 3rd edn. (eds Brian, W. J., Mahy, Marc, Van Regenmortel, H. V.) 374–380 (Academic Press, 2008).
Alfieri, A. A., Ribeiro, J., de Carvalho Balbo, L., Lorenzetti, E. & Alfieri, A. F. Dairy calf rearing unit and infectious diseases: Diarrhea outbreak by bovine coronavirus as a model for the dispersion of pathogenic microorganisms. Trop. Anim. Health Prod. 50, 1937–1940. https://doi.org/10.1007/s11250-018-1592-9 (2018).
Rodríguez-Prieto, V. et al. Evidence of shared bovine viral diarrhea infections between red deer and extensively raised cattle in south-central Spain. BMC Vet. Res. 12, 11. https://doi.org/10.1186/s12917-015-0630-3 (2016).
Waldeck, H. W. F. et al. Risk factors for introduction of bovine herpesvirus 1 (BoHV-1) into cattle herds: A systematic European literature review. Front. Vet. Sci. 8, 688935. https://doi.org/10.3389/fvets.2021.688935 (2021).
d’Offay, J. M., Fulton, R. W., Fishbein, M., Eberle, R. & Dubovi, E. J. Isolation of a naturally occurring vaccine/wild-type recombinant bovine herpesvirus type 1 (BoHV-1) from an aborted bovine fetus. Vaccine 37, 4518–4524. https://doi.org/10.1016/j.vaccine.2019.06.059 (2019).
Schroeder, R. J. & Moys, M. D. An acute upper respiratory infection of dairy cattle. J. Am. Vet. Med. Assoc. 125, 471–472 (1954).
Iscaro, C., Cambiotti, V., Petrini, S. & Feliziani, F. Control programs for infectious bovine rhinotracheitis (IBR) in European countries: An overview. Anim. Health Res. Rev. 22, 136–146. https://doi.org/10.1017/s1466252321000116 (2021).
Gaudino, M., Nagamine, B., Ducatez, M. F. & Meyer, G. Understanding the mechanisms of viral and bacterial coinfections in bovine respiratory disease: A comprehensive literature review of experimental evidence. Vet. Res. 53, 70. https://doi.org/10.1186/s13567-022-01086-1 (2022).
Hedayat, N. et al. Isolation and identification of bubaline herpesvirus 1 (BuHV-1) from latently infected water buffalo (Bubalus bubalis) from Iran. Trop. Anim. Health Prod. 52, 217–226. https://doi.org/10.1007/s11250-019-02007-9 (2020).
Hou, P., Xu, Y., Wang, H. & He, H. Detection of bovine viral diarrhea virus genotype 1 in aerosol by a real time RT-PCR assay. BMC Vet. Res. 16, 114–114. https://doi.org/10.1186/s12917-020-02330-6 (2020).
Antos, A., Miroslaw, P., Rola, J. & Polak, M. P. Vaccination failure in eradication and control programs for bovine viral diarrhea infection. Front. Vet. Sci. 8, 688911–688911. https://doi.org/10.3389/fvets.2021.688911 (2021).
Righi, C. et al. Validation of a commercial indirect ELISA kit for the detection of bovine alphaherpesvirus1 (BoHV-1)-specific glycoprotein e antibodies in bulk milk samples of dairy cows. Vet. Sci. https://doi.org/10.3390/vetsci9070311 (2022).
Maidana, S. S., Miño, S., Apostolo, R. M., De Stefano, G. A. & Romera, S. A. A new molecular method for the rapid subtyping of bovine herpesvirus 1 field isolates. J. Vet. Diagn. Invest. 32, 112–117. https://doi.org/10.1177/1040638719898692 (2020).
Pawar, S. S. et al. EvaGreen-based multiplex real-time PCR assay for rapid differentiation of wild-type and glycoprotein e-deleted bovine Herpesvirus-1 strains. Anim. Biotechnol. 28, 248–252. https://doi.org/10.1080/10495398.2016.1268620 (2017).
Walraph, J., Zoche-Golob, V., Weber, J. & Freick, M. Decline of antibody response in indirect ELISA tests during the periparturient period caused diagnostic gaps in Coxiella burnetii and BVDV serology in pluriparous cows within a Holstein dairy herd. Res. Vet. Sci. 118, 91–96. https://doi.org/10.1016/j.rvsc.2018.01.018 (2018).
Piepenburg, O., Williams, C. H., Stemple, D. L. & Armes, N. A. DNA detection using recombination proteins. PLoS Biol. 4, e204. https://doi.org/10.1371/journal.pbio.0040204 (2006).
Tan, M. et al. Recent advances in recombinase polymerase amplification: Principle, advantages, disadvantages and applications. Front. Cell Infect. Microbiol. 12, 1019071. https://doi.org/10.3389/fcimb.2022.1019071 (2022).
Singpanomchai, N. et al. Rapid detection of multidrug-resistant tuberculosis based on allele-specific recombinase polymerase amplification and colorimetric detection. PLoS One 16, e0253235. https://doi.org/10.1371/journal.pone.0253235 (2021).
Wang, J., Liu, L., Wang, J., Sun, X. & Yuan, W. Recombinase polymerase amplification assay—A simple, fast and cost-effective alternative to real time PCR for specific detection of feline Herpesvirus-1. PLoS One 12, e0166903. https://doi.org/10.1371/journal.pone.0166903 (2017).
Zhang, S. et al. Development of a directly visualized recombinase polymerase amplification-SYBR Green I method for the rapid detection of African swine fever virus. Front. Microbiol. 11, 602709. https://doi.org/10.3389/fmicb.2020.602709 (2020).
Zheng, Y. et al. RPA-SYBR Green I based instrument-free visual detection for pathogenic Yersinia enterocolitica in meat. Anal. Biochem. 621, 114157. https://doi.org/10.1016/j.ab.2021.114157 (2021).
Pang, J. et al. A real-time recombinase polymerase amplification assay for the rapid detection of Vibrio harveyi. Mol. Cell Probes 44, 8–13. https://doi.org/10.1016/j.mcp.2019.01.001 (2019).
Koo, K. M., Wee, E. J. & Trau, M. Colorimetric TMPRSS2-ERG gene fusion detection in prostate cancer urinary samples via recombinase polymerase amplification. Theranostics 6, 1415–1424. https://doi.org/10.7150/thno.15250 (2016).
Del Río, J. S., Svobodova, M., Bustos, P., Conejeros, P. & O’Sullivan, C. K. Electrochemical detection of Piscirickettsia salmonis genomic DNA from salmon samples using solid-phase recombinase polymerase amplification. Anal. Bioanal. Chem. 408, 8611–8620. https://doi.org/10.1007/s00216-016-9639-0 (2016).
Shahin, K. et al. Development of a recombinase polymerase amplification assay for rapid detection of Francisella noatunensis subsp. orientalis. PLoS One 13, e0192979. https://doi.org/10.1371/journal.pone.0192979 (2018).
Hou, P., Wang, H., Zhao, G., He, C. & He, H. Rapid detection of infectious bovine Rhinotracheitis virus using recombinase polymerase amplification assays. BMC Vet. Res. 13, 386. https://doi.org/10.1186/s12917-017-1284-0 (2017).
Ullah, N., Assawakongkarat, T., Akeda, Y. & Chaichanawongsaroj, N. Detection of extended-spectrum β-lactamase-producing Escherichia coli isolates by isothermal amplification and association of their virulence genes and phylogroups with extraintestinal infection. Sci. Rep. 13, 12022. https://doi.org/10.1038/s41598-023-39228-w (2023).
Hassan, M. A. et al. Simultaneous detection of Theileria annulata and Theileria orientalis infections using recombinase polymerase amplification. Ticks Tick-Borne Dis. 9, 1002–1005. https://doi.org/10.1016/j.ttbdis.2018.03.028 (2018).
Zhai, J. et al. Detection of Neisseria gonorrhoeae and Chlamydia trachomatis infections in pregnant women by multiplex recombinase polymerase amplification. PLoS One 16, e0251119. https://doi.org/10.1371/journal.pone.0251119 (2021).
Conrad, C. C. et al. A sensitive and accurate recombinase polymerase amplification assay for detection of the primary bacterial pathogens causing bovine respiratory disease. Front. Vet. Sci. 7, 208. https://doi.org/10.3389/fvets.2020.00208 (2020).
Socha, W., Larska, M., Rola, J. & Bednarek, D. Occurrence of bovine coronavirus and other major respiratory viruses in cattle in Poland. J. Vet. Res. 66, 479–486. https://doi.org/10.2478/jvetres-2022-0059 (2022).
Benavides, B. et al. Quantitative risk assessment of introduction of BVDV and BoHV-1 through indirect contacts based on implemented biosecurity measures in dairy farms of Spain. Prev. Vet. Med. 188, 105263. https://doi.org/10.1016/j.prevetmed.2021.105263 (2021).
Diao, N. C. et al. Prevalence of bovine viral diarrhea virus (BVDV) in yaks between 1987 and 2019 in mainland China: A systematic review and meta-analysis. Microb. Pathog. 144, 104185. https://doi.org/10.1016/j.micpath.2020.104185 (2020).
Yang, S. et al. A lateral flow dipstick combined with reverse transcription recombinase polymerase amplification for rapid and visual detection of the BVDV and BPIV3. J. Virol. Methods 299, 114343. https://doi.org/10.1016/j.jviromet.2021.114343 (2022).
Marin, M. et al. Distinctive features of bovine alpha herpesvirus types 1 and 5 and the virus-host interactions that might influence clinical outcomes. Arch. Virol. 165, 285–301. https://doi.org/10.1007/s00705-019-04494-5 (2020).
Yu, Z. et al. Development of a droplet digital PCR assay to detect bovine alpha herpesvirus 1 in bovine semen. BMC Vet. Res. 18, 125. https://doi.org/10.1186/s12917-022-03235-2 (2022).
Petrini, S., Iscaro, C. & Righi, C. Antibody responses to bovine alphaherpesvirus 1 (BoHV-1) in passively immunized calves. Viruses https://doi.org/10.3390/v11010023 (2019).
Srisrattakarn, A. et al. Direct detection of methicillin-resistant in Staphylococcus spp. in positive blood culture by isothermal recombinase polymerase amplification combined with lateral flow dipstick assay. World J. Microbiol. Biotechnol. 36, 162. https://doi.org/10.1007/s11274-020-02938-8 (2020).
Du, X.-J., Zang, Y.-X., Liu, H.-B., Li, P. & Wang, S. Rapid detection of Staphylococcus aureus via recombinase polymerase amplification combined with lateral flow strip. Food Anal. Methods 11, 2296–2306. https://doi.org/10.1007/s12161-018-1200-7 (2018).
Crannell, Z. A. et al. Nucleic acid test to diagnose cryptosporidiosis: lab assessment in animal and patient specimens. Anal. Chem. 86, 2565–2571. https://doi.org/10.1021/ac403750z (2014).
Moore, M. D. & Jaykus, L.-A. Recombinase polymerase amplification: A promising point-of-care detection method for enteric viruses. Future Virol. 12, 421–429. https://doi.org/10.2217/fvl-2017-0034 (2017).
Peltzer, D., Tobler, K., Fraefel, C., Maley, M. & Bachofen, C. Rapid and simple colorimetric loop-mediated isothermal amplification (LAMP) assay for the detection of bovine alphaherpesvirus 1. J. Virol. Methods 289, 114041. https://doi.org/10.1016/j.jviromet.2020.114041 (2021).
Socha, W., Rola, J., Urban-Chmiel, R. & Żmudziński, J. F. Application of loop-mediated isothermal amplification (LAMP) assays for the detection of bovine herpesvirus 1. Pol. J. Vet. Sci. 20, 619–622. https://doi.org/10.1515/pjvs-2017-0078 (2017).
Silva, G. et al. Rapid detection of potyviruses from crude plant extracts. Anal. Biochem. 546, 17–22. https://doi.org/10.1016/j.ab.2018.01.019 (2018).
Acknowledgements
Thanks to Ningxia University for providing the experimental platform.
Funding
This work was supported by the National Natural Science Foundation of China (grant number: 32130104), the Key Research, Development Program of Ningxia Hui Autonomous Region (grant number 2021BEF02028) and Ningxia Natural Science Foundation (2022AAC03077) .
Author information
Authors and Affiliations
Contributions
JLL participated in study design, method establishment, data organization, manuscript writing and revision. ZG collaborated in data preparation, analysis and interpretation. WP, NXX, LQ, ZSN and GWF participated in sample collection and preparation, article editing and proofreading. LY did conceptualization, funding acquisition, project management, resources.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, L., Zhang, G., Wang, P. et al. Simultaneous detection of bovine viral diarrhea virus (BVDV) and bovine herpesvirus 1 (BoHV-1) using recombinase polymerase amplification. Sci Rep 14, 10169 (2024). https://doi.org/10.1038/s41598-024-56869-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-56869-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
