
Abstract
Coxiella burnetii, a zoonotic pathogen, is the causative agent of Q fever, an endemic disease in Iran. However, there is currently a lack of available data on the genotypes of C. burnetii in the country. Here, we typed 26 C. burnetii isolates detected in milk, abortion, cotylodon, and cardiac valve samples from various geographical areas and hosts (7 cattle, 8 goats, 10 sheep, and 1 human) using Multilocus Variable Number Tandem Repeat Analysis (MLVA/VNTR) with five loci:ms24, ms27, ms28, ms33, and ms34. As IS1111 was observed to be spontaneously inserted in locus ms23 across all of our examined C. burnetii samples, five loci were employed for MLVA/VNTR genotyping. Among the 26 C. burnetii strains, 22 distinct genotypes (A–V) were identified in the discriminative loci. In silico analysis categorized Iranian C. burnetii strains into five genomic groups along with seven singletons, representing 11 exiting clonal complexes worldwide. Clusters 10 and 11 exclusively consisted of Iranian samples. These findings revealed high genotyping diversity among C. burnetii isolates in Iran. The genotypes circulating in Iran differed significantly from those found in other regions worldwide. To gain a comprehensive understanding of Q fever epidemiology in Iran, it is crucial to conduct large-scale studies that assess the distribution of C. burnetii genotypes across different geographical areas, hosts, and sources.
Similar content being viewed by others
Introduction
Q fever is a zoonotic disease caused by Coxiella burnetii, an obligate intracellular bacterium. While domestic ruminants, such as cattle, sheep, and goats, are the main reservoirs of C. burnetii, infections in these animals may be asymptomatic1. However, during outbreaks, goats may experience significant abortion rates of up to 90%2. The primary transmission route of C. burnetii to humans is through contaminated aerosols and dust particles. However, rare cases of transmission of infection to humans through ingestion of raw milk or dairy products, sexual contact, and blood transfusion have also been reported3,4.
Q fever can manifest in humans as either asymptomatic or self-limiting5, with acute cases resembling flu-like symptoms. In some instances, clinical manifestations of acute Q fever may include hepatitis and atypical pneumonia6, while chronic Q fever can lead to life-threatening endocarditis3.
Annually, there are reports of infections caused by C. burnetii in humans and animals worldwide3. In Europe, seroprevalence studies estimate the occurrence of C. burnetii in humans to be between 2 and 10%7. According to a meta-analysis study conducted in Iran in 2017, the overall seroprevalence of phase I and II IgG antibodies in humans was found to be 19.8% and 32.86%, respectively1. The prevalence of acute Q fever among suspected patients in northern and northwestern Iran was reported to be 13.8% and 5.37% in 2017 and 2019, respectively8,9. Q fever infective endocarditis was diagnosed in 30.77% of patients with culture-negative endocarditis in Iran from 2016 to 201810. In addition, there have been several reports of C. burnetii infections in domestic livestock, wildlife, ticks, milk, and dairy product1,11,12. However, limited data exists on C. burnetii molecular typing in Iran. A recent study showed the circulation of five C. burnetii genotypes among Iranian domestic ruminants using the multi-spacer sequence typing (MST) method13.
Given the wide geographical distribution, diverse reservoirs, transmission routes, and clinical features of C. burnetii, studying its epidemiology presents a complex challenge. Therefore, molecular characterization of C. burnetii strains can provide valuable epidemiological data14,15,16. Comparative analysis of C. burnetii genotypes can help identify sources of infection in humans and animals, classify circulating strains, and contribute to infection control and prevention17. Multilocus Variable Number Tandem Repeat Analysis (MLVA/VNTR) genotyping, based on Variable Number of Tandem Repeats (VNTR), offers a helpful approach for C. burnetii genotyping. This method was first designed in 2006 for typing C. burnetii isolates, resulting in the identification of 36 different genotypes among 42 C. burnetii isolates14. The first panel of MLVA/VNTR genotyping contains eleven loci with repeat units equal to or longer than nine base pairs (bps), while the second panel includes six loci with repeat units of six or seven base pairs (bps) that require more advanced separation methods such as capillary electrophoresis14,18.
As Q fever is endemic in Iran and comprehensive data on genotyping of circulating C. burnetii strains is lacking, molecular genotyping using the MLVA/VNTR method can significantly contribute to understanding the molecular epidemiology of Q fever in Iran. Therefore, the present study aimed to address this gap by conducting molecular genotyping of selected C. burnetii isolates in Iran using the MLVA/VNTR method with the second panel.
Material and methods
Samples
The 26 C. burnetii-positive samples previously identified in Iran10,19,20,21 were selected for genotyping using the Dutch six-locus MLVA panel22. These positive samples consisted of one human sample (cardiac valve from a Q fever endocarditis case) and 25 animal samples. All samples included in this study were confirmed using a quantitative real-time PCR (qPCR) assay targeting IS1111 of C. burnetii23. The characteristics of included samples and their quantification cycle (Cq) values obtained via qPCR are summarized in Table 1.
MLVA/VNTR capillary electrophoresis genotyping
Since six known loci or VNTRs, namely ms23, ms24, ms27, ms28, ms33, and ms34, were utilized for genotyping C. burnetii using capillary electrophoresis in established C. burnetii MLVA/VNTR studies, we also initially targeted these six loci with the previously designated primers22,24. However, during gel electrophoresis of the amplified ms23 locus, an unusual fragment with a length of 1500 bp was observed in all 26 samples analyzed. Sequencing analysis of the PCR product from this locus (ms23) for all samples revealed that the IS1111 insertion sequence was present at this site, causing an increase in its size. Therefore, the amplification of the ms23 locus was unsuccessful for the MLVA/VNTR study. Considering this limitation, we excluded it from the analysis and utilized five loci instead of six. Reverse primers for the ms23, ms33, and ms34 loci were labeled with the 6-FAM fluorescent color, while reverse primers for the ms24, ms27, and ms28 loci were labeled unilaterally with carboxy 4, 7, 2', 4', 5', 7' hexachloro fluorescein (HEX) -6. The PCR reaction was performed using the commercial TEMPase Hot Start 2× Master Mix BLUE (Ampliqon, Odense M, Denmark). Each reaction contained 5 μl of extracted DNA, 12.5 μl of Mastermix, 400 mmol of each primer, and distilled water added to achieve a final volume of 25 μl. The standard strain used in this study was the Nine Mile RSA493 C. burnetii strain.
The PCR temperature program consisted of an initial denaturation step at 95 °C for 15 min, followed by 40 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 60 s, extension at 72 °C for 1 min, and a final extension step at 72 °C for 10 min. To confirm the presence of the desired band and the absence of non-specific bands, the PCR product for each locus was separated by size using a 2% agarose gel. Following successful amplification, capillary electrophoresis was carried out to determine the exact size of each locus. The purified PCR products were obtained using a PCR product purification kit (Kawsar biotech, Tehran, Iran) and were further diluted at ratios of 1: 5, 1: 10, and 1: 100. Then, 2 μl of each diluted sample was added to a solution containing 10.5 μl of formide and 0.5 μl of standard marker size 560 (Applied Biosystems, Foster City, USA). The mixture was thoroughly mixed, denatured at 95 °C for 30 s, and then placed on ice for 2 min. Finally, the samples were run on a Genetic Analyzer capillary sequencer model 3130x (Applied Biosystems, Foster City, USA) using POP7 polymer.
The data obtained from capillary electrophoresis were analyzed using GeneMarker software, version 1.6 (Pennsylvania, USA), regarding the standard marker size and the fluorescent dye for each locus.
Determine the number of repeats for each VNTR locus
The number of repeats for each VNTR locus was determined by comparing the sizes of the fragments obtained with those obtained from the reference strain Nine Mile RSA 493 in the same run. Based on in silico analysis, the established genotype of the strain was found to have 9, 27, 4, 6, 9, and 5 repeats for marker loci Ms23, Ms24, Ms-27, Ms28, Ms33, and Ms34, respectively.
The following formula, which replaces the size and number of repeats in each locus of the standard Nine Mile RSA493 C. burnetii strain, was used to determine the number of repetitions for each VNTR locus in the samples25.
VNTR data analysis
The discriminatory power of each selected VNTR locus was calculated by the Hunter and Gaston discriminatory index (HGDI)26.
The MLVA/VNTR data of the examined samples were analyzed using the online MLVAbank for Microbes Genotyping database (http://microbesgenotyping.i2bc.paris-saclay.fr/). The profiles of each sample were compared with previously registered strains in the database. Genotypes that were not previously registered in the database were considered new genotypes of C. burnetii. The data from five capillary loci in this study were compared against the information available in the databases.
The phylogenetic analysis was conducted using the advanced Bionumerics 6.6 software package (Applied Maths, Sint-Martens-Latem, Belgium). The comparison of the position of C. burnetii strains with other data recorded in the database was determined using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) and Minimum Spanning Tree algorithms. Genomic grouping for C. burnetii was performed using cluster analysis for MLVA/VNTR capillary electrophoresis data.
Ethics
This study, including the proposal, all experimental protocols, and consent procedure, was approved by the Ethics Committee for Biomedical Research of Tarbiat Modares University (Ethic Code: IR.TMU.REC.1395.510). All methods were carried out in accordance with the relevant guidelines and regulations of the Ethics Committee for Biomedical Research of Tarbiat Modares University. The related methods are reported following the ARRIVE guidelines (https://arriveguidelines.org).
We used the samples available in our biobank, which were collected during our previous studies10,19,20,21. Therefore, the requirement for an informed consent statement was waived.
Results
MLVA/VNTR genotyping with the five-locus panel using capillary electrophoresis
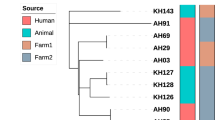
Finally, 22 genotypes (A to V) of C. burnetii were observed among the studied samples in this study (Table 2). The genotype of the human heart valve sample (Sample Q34) was relatively determined as it was amplified in only three out of the five loci tested. However, it showed the closest similarity to the standard control strain (Nine Mile RSA493 C. burnetii). Furthermore, genotypes C (samples AG2 and AS3), E (samples MS51 and MS52), F (samples MG103 and MS16), and S (samples 9898 and MB47) included two samples with similar allelic profiles. Based on genotyping results of 27 samples for the panel of five tested loci, the overall HGDI value of the MLVA/VNTR genotyping method was estimated at 0.740. The HGDI value for a single locus ranged from 0.493 to 0.865. The HGDI value for ms24 (10 alleles), ms27(5 alleles), ms28(8 alleles), ms33 (10 alleles), and ms34 (8 alleles) was 0.836, 0.493, 0.729, 0.752, and 0.865, respectively. The UPGMA analyses of samples in this study are presented in Fig. 1.

Dendogram designed of Coxiella burnetii based on UPGMA analysis of MLVA/VNTR 5 loci (ms24, ms27, ms28, ms33 and ms34).
The Minimum Spanning Tree algorithm analysis of C. burnetii samples in Iran, compared to world strains (based on data recorded in the online MLVAbank for Microbes Genotyping database), is shown in Fig. 2.

MLVA/VNTR analysis of Coxiella burnetii using the Minimum Spanning Tree algorithm for MLVA/VNTR 5 loci (ms24, ms27, ms28, ms33 and ms34) data obtained from the online MLVAbank for Microbes Genotyping database (http://microbesgenotyping.i2bc.paris-saclay.fr/). The origin of the samples is represented by different colors. Our samples are indicated with an arrow.
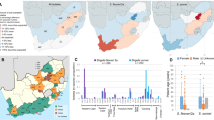
In silico cluster analysis
Using cluster analysis of MLVA/VNTR capillary electrophoresis data, 11 genomic and clonal complex groups were identified for C. burnetii (Fig. 3). Iranian C. burnetii strains were categorized into five distinct genomic groups (clusters 1, 2, 3, 10, and 11) as well as seven singletons. The majority of Iranian samples (nine strains) were classified in cluster 3. Clusters 11 (three samples) and 10 (two samples) exclusively consisted of Iranian samples. Cluster 2 represented the largest genome group, with two samples from Iran. Additionally, cluster 1, the second-largest cluster, featured a sample (MB92) from Iran.

Genomic grouping (cluster 1–11) of Coxiella burnetii using the Minimum Spanning Tree algorithm based on data obtained from the online MLVAbank for Microbes Genotyping database (http://microbesgenotyping.i2bc.paris-saclay.fr/)and the clustering of genotypes.
Discussion
Q fever has emerged as a significant global public health concern due to various factors, including the wide range of hosts that C. burnetii can infect, its transmission through aerosols, its low infectious dose, and its ability to survive in the environment for long periods. Early studies demonstrated that C. burnetii strains collected from different regions and sources exhibit differences in virulence and signs of disease, which can be attributed to the diversity of C. burnetii genotypes17,27,28. Therefore, genotyping provides valuable insights into the genotype of circulating C. burnetii strains and helps to identify sources. These capabilities make genotyping an essential tool for the control of C. burnetii outbreaks29. The present study reported the first genotyping of C. burnetii strains in Iran using the MLVA/VNTR method with five different loci (Ms24, Ms27, Ms28, Ms33, and Ms34) and capillary electrophoresis. Unfortunately, one locus (Ms23) was excluded from genotyping due to the non-successful amplification caused by an IS1111 insertion inside it. This insertion has also been reported in some strains of C. burnetii in previous studies30,31. In addition, out of the five loci tested, only three were amplified in sample Q34 (human heart valve), resulting in a partially determined genotype. The genotype of this sample was relatively close to the cotyledon sample derived from a sheep (AS43).
Using this genotyping panel, we identified 22 different genotypes (A to V) among the 26 livestock and human samples selected from six cities, indicating a high level of diversity in genotyping C. burnetii genotypes in Iran. This is not surprising, as previous studies have also shown a diverse range of genotypes associated with different hosts or sources of C. burnetii infection17,27,28.
Among the samples derived from sheep, goats, and cattle, we identified nine, eight, and six different genotypes, respectively. However, two genotypes (F and C) were shared between samples from sheep and goats, suggesting some commonality in the genotypes of C. burnetii infecting these host species. Several studies conducted in multiple countries have discovered various MLVA/VNTR genotypes of C. burnetii that infect dairy cattle herds29,32,33. For example, in Germany, 32 distinct genotypes were identified among 104 C. burnetii strains derived from cattle and sheep34. In Italy, 23 different genotypes were reported among 34 C. burnetii isolates infecting dairy cattle products35. Similar diversity was observed in C. burnetii strains isolated from human clinical and animal samples in other countries like Portugal36 and Hungary37. In the Netherlands, identical genotypes between humans and goats showed that goats were likely sources of human infection38. These studies highlight the diverse genotypic variations of C. burnetii isolates across different hosts and regions, emphasizing the importance of comprehensive genotyping methods in understanding the epidemiology of this pathogen. In addition to host-related genetic diversity, we also observed genetic diversity based on geographic distribution within Iran. Each city in the present study had different genotypes, except for two in Qom (genotype E) and two in Tehran (genotype C). The genotypes identified in this study were specific to different regions and the country. These variations may be influenced by different climate and environmental conditions in each region, as the epidemiology of tick-borne diseases has been shown to be significantly affected by environmental changes resulting from both the climate emergency and human activities39.
The cluster analysis of available C. burnetii MLVA/VNTR capillary electrophoresis data revealed the presence of 11 (1–11) genomic and clonal complex groups recorded in the word. Furthermore, this analysis revealed that C. burnetii strains in Iran were classified into five different genomic groups, with two clusters (10 and 11) exclusively comprised of strains from Iran. These findings highlight the distinctiveness of C. burnetii genotypes in Iran compared to those reported from other parts of the world. However, the limitations of this study include the relatively small sample size, the inability to amplify and analyze the ms23 locus (this reduced the MLVA/VNTR panel to 5 loci instead of 6, limiting resolution), and the lower resolution offered by MLVA compared to other advanced genotyping techniques like whole-genome sequencing. Future studies with larger sample sizes and more comprehensive genotyping methods are needed to further enhance our understanding of the molecular epidemiology of C. burnetii in Iran.In conclusion, the strains described in the present study, exhibiting various host tropisms and geographical origins, have revealed the diverse genetic profiles of C. burnetii strains in Iran. Genotyping analysis revealed significant differences between circulating strains of C. burnetii in Iran compared to strains from other regions. This information provides valuable insights into the molecular epidemiology of C. burnetii in Iran and emphasizes the need for comprehensive surveillance, prevention, and treatment strategies for Q fever in the country.
Data availability
Data supporting the findings of this study are available within the article and can be obtained from the corresponding author upon request. The MLVA/VNTR genotyping data for C. burnetii in the present study have been deposited in MLVAbank for Microbes Genotyping (https://microbesgenotyping.i2bc.paris-saclay.fr/).
References
Mohabbati Mobarez, A., Bagheri Amiri, F. & Esmaeili, S. Seroprevalence of Q fever among human and animal in Iran; A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 11, e0005521 (2017).
Anastácio, S., de Sousa, S. R., Saavedra, M. J. & da Silva, G. J. Role of Goats in the Epidemiology of Coxiella burnetii. Biology (Basel). 11, 1703 (2022).
Eldin, C. et al. From Q fever to Coxiella burnetii infection: a paradigm change. Clin. Microbiol. Rev. 30, 115–190 (2017).
Maurin, M. & Raoult, D. Q fever. Clin. Microbiol. Rev. 12, 518–553 (1999).
Raoult, D., Marrie, T. & Mege, J. Natural history and pathophysiology of Q fever. Lancet Infect. Dis. 5, 219–226 (2005).
Slok, E. N. et al. Estimation of acute and chronic Q fever incidence in children during a three-year outbreak in the Netherlands and a comparison with international literature. BMC Res. Notes. 8, 1–7 (2015).
Georgiev, M. et al. Q fever in humans and farm animals in four European countries, 1982 to 2010. Euro Surveill. 18, 20407 (2013).
Esmaeili, S., Golzar, F., Ayubi, E., Naghili, B. & Mostafavi, E. Acute Q fever in febrile patients in northwestern of Iran. PLoS Negl. Trop. Dis. 11, e0005535 (2017).
Ghasemian, R., Mostafavi, E., Esmaeili, S., Arabsheybani, S. & Davoodi, L. Epidemiologic investigation of acute Q fever in North of Iran. J. Maz. Univ. Med. Sci. 29, 100–106 (2019).
Moradnejad, P. et al. Q fever endocarditis in Iran. Sci Rep. 9, 1–7 (2019).
Fard, S. R. & N., Ghashghaei, O., Khalili, M. & Sharifi, H.,. Tick diversity and detection of Coxiella burnetii in tick of small ruminants using nested Trans PCR in Southeast Iran. Trop. Biomed. 33, 506–511 (2016).
Esmaeili, S., Mobarez, A. M., Khalili, M., Mostafavi, E. & Moradnejad, P. Molecular prevalence of Coxiella burnetii in milk in Iran: A systematic review and meta-analysis. Trop. Anim. Health Prod. 51, 1345–1355 (2019).
Mohabati Mobarez, A. et al. Genetic diversity of Coxiella burnetii in Iran by multi-spacer sequence typing. Pathogens. 11, 1175 (2022).
Arricau-Bouvery, N. et al. Molecular characterization of Coxiella burnetii isolates by infrequent restriction site-PCR and MLVA typing. BMC Microbiol. 6, 1–14 (2006).
Yanmaz, B. & Ozgen, E. K. Detection of Coxiella burnetii and characterisation by multiple-locus variable-number tandem repeat analysis in bovine bulk tank milk samples. Vet. Med. (Praha). 68, 185 (2023).
Truong, A.-T. et al. Genotyping of Coxiella burnetii from cattle by multispacer sequence typing and multiple locus variable number of tandem repeat analysis in the Republic of Korea. Genes. 13, 1927 (2022).
Hemsley, C. M., Essex-Lopresti, A., Norville, I. H. & Titball, R. W. Correlating genotyping data of Coxiella burnetii with genomic groups. Pathogens. 10, 604 (2021).
Svraka, S., Toman, R., Skultety, L., Slaba, K. & Homan, W. L. Establishment of a genotyping scheme for Coxiella burnetii. FEMS Microbiol. Lett. 254, 268–274 (2006).
Mohabati Mobarez, A., Khalili, M., Mostafavi, E. & Esmaeili, S. Molecular detection of Coxiella burnetii infection in aborted samples of domestic ruminants in Iran. PLoS ONE. 16, e0250116 (2021).
Mobarez, A. M., Mostafavi, E., Khalili, M. & Esmaeili, S. Identification of Coxiella burnetii in raw milk of livestock animal in Iran. Int. J. Microbiol. 2021 (2021).
Esmaeili, S., Mobarez, A. M., Khalili, M. & Mostafavi, E. High prevalence and risk factors of Coxiella burnetii in milk of dairy animals with a history of abortion in Iran. Comp. Immunol. Microbiol. Infect. Dis. 63, 127–130 (2019).
Tilburg, J. J. et al. Genotypic diversity of Coxiella burnetii in the 2007–2010 Q fever outbreak episodes in The Netherlands. J. Clin. Microbiol. 50, 1076–1078 (2012).
Schneeberger, P. M. et al. Real-time PCR with serum samples is indispensable for early diagnosis of acute Q fever. Clin. Vaccine Immunol. 17, 286–290 (2010).
Klaassen, C. H., Nabuurs-Franssen, M. H., Tilburg, J. J., Hamans, M. A. & Horrevorts, A. M. Multigenotype Q fever outbreak, the Netherlands. J. Emerg. Infect. Dis. 15, 613 (2009).
Szymańska-Czerwińska, M., Jodełko, A., Zaręba-Marchewka, K. & Niemczuk, K. Shedding and genetic diversity of Coxiella burnetii in Polish dairy cattle. PLoS ONE. 14, e0210244 (2019).
Hunter, P. R. & Gaston, M. A. Numerical index of the discriminatory ability of typing systems: An application of Simpson’s index of diversity. J. Clin. Microbiol. 26, 2465–2466 (1988).
Glazunova, O. et al. Coxiella burnetii genotyping. J. Emerg. Infect. Dis. 11, 1211 (2005).
Long, C. M., Beare, P. A., Cockrell, D. C., Larson, C. L. & Heinzen, R. A. Comparative virulence of diverse Coxiella burnetii strains. Virulence. 10, 133–150 (2019).
Mioni, Md. S. R. et al. New genotypes of Coxiella burnetii circulating in Brazil and Argentina. Pathogens. 9, 30 (2019).
Sidi-Boumedine, K. et al. Impact of IS1111 insertion on the MLVA genotyping of Coxiella burnetii. Microbes Infect. 17, 789–794 (2015).
Di Domenico, M. et al. Genetic diversity of Coxiella burnetii in domestic ruminants in central Italy. BMC Vet. Res. 14, 1–7 (2018).
Hemsley, C. M. et al. MLVA and com1 genotyping of Coxiella burnetii in farmed ruminants in Great Britain. Vet Microbiol. 277, 109629 (2023).
Bauer, B. U. et al. Multispecies Q fever outbreak in a mixed dairy goat and cattle farm based on a new bovine-associated genotype of Coxiella burnetii. Vet. Sci. 8, 252 (2021).
Frangoulidis, D. et al. Molecular analysis of Coxiella burnetii in Germany reveals evolution of unique clonal clusters. Int. J. Med. Microbiol. 304, 868–876 (2014).
Ceglie, L. et al. Molecular characterization by MLVA of Coxiella burnetii strains infecting dairy cows and goats of north-eastern Italy. Microbes Infect. 17, 776–781 (2015).
Santos, A. S. et al. Genotypic diversity of clinical Coxiella burnetii isolates from Portugal based on MST and MLVA typing. Int J Med Microbiol. 302, 253–256 (2012).
Sulyok, K. M. et al. Genotyping of Coxiella burnetii from domestic ruminants and human in Hungary: Indication of various genotypes. BMC Vet. Res. 10, 1–6 (2014).
Tilburg, J. J. et al. Epidemic genotype of Coxiella burnetii among goats, sheep, and humans in the Netherlands. J. Emerg. Infect. Dis. 18, 887 (2012).
Ebani, V. V. et al. Molecular survey on the presence of arthropod-borne bacteria and protozoans in roe deer (Capreolus capreolus) and ticks from Central Italy. Acta Trop. 233, 106586 (2022).
Acknowledgements
We would like to express our gratitude to Tarbiat Modares University, Pasteur Institute of Iran, the Centre for Communicable Diseases Control in the Ministry of Health, and the Iranian National Scientific Foundation for their financial support of this study. We would also like to express our gratitude to Dr. Kayvan Tadayon (Razi Vaccine & Serum Research Institute) for his technical support in the phylogenetic analysis of this study.
Funding
This work was financially supported by Tarbiat Modares University (Tehran, Iran), Pasteur Institute of Iran, Centre for Communicable Diseases Control in the Ministry of Health (Grant 810), and the Iranian National Scientific Foundation (INSF; Contracted No. 91004716). The funders had no role in study design, survey conduct, or results analysis.
Author information
Authors and Affiliations
Contributions
A.M.M., M.K., and S.E. participated in the conception, validation, and design of the study. A.M.M., E.M., and S.E. acquired the funds. S.E. performed experiments. S.E. and E.M. analyzed data. N.B. involved in writing—original draft preparation. N.B. reviewed, edited, and had scientific suggestions for a better presentation of the manuscript. All authors have read and agreed to the published version of the manuscript. A.M.M. and N.B. have made equal contributions to this study and should be considered as co-first authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohabati Mobarez, A., Baseri, N., Khalili, M. et al. Genotyping and phylogenetic analysis of Coxiella burnetii in domestic ruminant and clinical samples in Iran: insights into Q fever epidemiology. Sci Rep 13, 20374 (2023). https://doi.org/10.1038/s41598-023-47920-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-47920-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
