
Abstract
Three hundred and forty-four tick samples were collected from vegetation at Taksin Maharat National Park, Tak province, northwestern Thailand. They were morphologically identified and molecularly confirmed by 16S rRNA and COI genes as Dermacentor laothaiensis (n = 105), D. steini (n = 139), and D. auratus (n = 100). These ticks were examined for the spotted fever group rickettsiae (SFGRs) using PCR and DNA sequencing of six genes; 17-kDa, gltA, 16S rRNA, ompA, ompB, and sca4. Of these ticks, 6.10% (21/344) gave positive results for the presence of SFGRs. Phylogenetic analyses of the SFGRs clearly indicated that a novel genotype assigned as Candidatus Rickettsia takensis was detected in D. laothaiensis (19/105) and at lesser frequency in D. steini (1/139). Furthermore, Candidatus Rickettsia laoensis was also found at a low frequency in D. auratus (1/100), the first record in Thailand. Although, the pathogenicities of these SFGRs remain unknown, our findings suggest potential risks of SFGRs being transmitted via ticks near the border between Thailand and Myanmar, a gateway of daily migrations of local people and visitors both legal and illegal.
Similar content being viewed by others

Introduction
Spotted fever group rickettsiae (SFGRs) belonging to the Order Rickettsiales are obligate intracellular Gram-negative bacteria; i.e. the bacteria need to associate with eukaryotic cells1. The link between SFGRs and emerging and re-emerging zoonosis infectious vector-borne diseases, is well recognized and studied worldwide2,3. At least thirty species of SFGRs have been reported over the past 35 years. Of these, 21 species have been identified as pathogens and others are of unknown pathogenicity2,4,5. Many species of rickettsiae were identified in ticks long before they were recognized as pathogenic agents in humans because the amount of Rickettsia DNA in arthropods is higher than in human blood3. Therefore, studies on species diversity of potential tick vectors of SFGRs are crucial to determine the presence or absence of any Rickettsia pathogens causing rickettsioses in humans.
At least five species of SFGRs (R. conorii, R. helvetica, R. honei, R. japonica, and R. rhipicephali) have been reported infecting humans in Southeast Asia1,5 although the incidence of spotted fever group rickettsioses varies across the region. The first four species listed above have been found in patients in Thailand6,7,8,9,10,11. Pathogenic Rickettsia species and other known rickettsial bacteria are associated with arthropod vectors, especially with hard ticks: these include Ixodes granulatus, Dermacentor atrosignatus, Dermacentor spp., Haemaphysalis hystricis, Haemaphysalis spp., and Amblyomma spp.12,13,14,15,16,17,18. In 2016, three divergent genotypes of SFGRs, namely Ca. R. laoensis and Ca. R. khammouanensis infecting Haemaphysalis sp., and Ca. R. mahosotii infecting A. testudinarium and Haemaphysalis sp. were reported in Laos but their pathogenicities were unknown16.
Identification of novel species of tick-borne rickettsiae has been greatly facilitated by DNA technology, especially gene sequence-based methods. Thus, molecular techniques are beneficial for the classification of Rickettsia as demonstrated by the description of the cultured R. peacockii based on genes 16S rDNA, gltA, and ompA sequence similarity, which was detected in D. andersoni from western Montana, USA19. Furthermore, a detailed study of Rickettsia using nucleotide sequence-based analysis of 5 genes (16S rRNA, gltA, ompA, ompB, and sca4) for classification of genus or species of SFGRs was proved to be a discriminative method for identification of the cultured R. heilongjiangensis found in D. silvarum from Heilongjiang, China20. In addition, Diop et al. used multilocus sequence typing (MLST) scheme methods to identify a novel Rickettsia species, R. fournieri, detected in Argas lagenoplastis ticks from Queensland, Australia21. Recently, Li et al. employed MLST to describe a new SFGR, Ca. R. xinyangensis, detected both in a patient and in Haemaphysalis longicornis ticks from Xinyang, China22.
In the present study, MLST was applied to evaluate the genetic polymorphisms and phylogenetic relationships of SFGRs detected in Dermacentor ticks residing in vegetation at Taksin Maharat National Park, Tak province, northwestern Thailand. The results indicated a novel genotype of Rickettsia, putatively named Candidatus Rickettsia takensis, detected in D. laothaiensis and D. steini ticks. The present report also provides the first record of Ca. R. laoensis infecting D. auratus in Thailand. We also present molecular evidence for species identifications of D. laothaiensis, D. steini, and D. auratus.
Results
Tick species identification
A total of 344 tick samples from the study area comprised three morphological species of Dermacentor ticks. These were D. laothaiensis (n = 105: 36 males, 69 females), D. steini (n = 139: 68 males, 71 females), and D. auratus (n = 100: 42 males, 58 females) (Fig. 1a). The morphology of D. laothaiensis was observed to be very similar to D. steini, and both were clearly different from D. auratus (Fig. 1b–j). D. steini showed closely spaced external and internal spurs on coxa-I, a distinct narrow dark border stripe on the densely punctuated scutum or pseudoscutum, and a narrowly to broadly V-shaped aperture of the female genital structure. D. laothaiensis, otherwise similar in morphology to D. steini, differed in having an indistinct broad hazy central brown stripe on the scutum or pseudoscutum, moderately dense large punctations, and very narrowly V-shaped genital aperture. D. auratus exhibited widely spaced external and internal spurs on coxa-I, a distinct narrow line through the center of scutum/ pseudoscutum, and a U-shaped genital aperture; Fig. 1b–j. Phylogenetic analysis results based on 16S rRNA and COI genes confirmed the existence of these 3 related species (Figs. 2a, c and S1). The pairwise diagram for these tick species based on 16S rRNA gene (Fig. 2b) demonstrated that D. laothaiensis had 91%, 91.8%, and 92% similarity compared with D. steini from Thailand (ON705029), D. auratus from Thailand (ON705025), and D. steini from Malaysia (MK296405), respectively. Their corresponding total numbers of differentiation positions of nucleotide base sequences alignments were 36 (27 bases/ 9 gaps), 33 (31 bases/ 2 gaps), and 32 (29 bases/ 3 gaps), respectively. Thailand D. steini was 91% similar to D. auratus from Thailand (ON705025) and 95.8% similar to D. steini from Malaysia (MK296405). It differed from both taxa in positions of nucleotide base sequences alignments by 36 (31 bases/ 5 gaps), and 17 (15 bases/ 2 gaps), respectively. Our D. auratus tick sample, showed a 99.5% match with both a D. auratus specimen originating from Malaysia (MT914184) and one other from Thailand (KC170746). Their total numbers of differentiation positions of nucleotide base sequences alignments were 2 (2 bases/ 0 gap) and 2 (1 base/ 1 gap), respectively. Likewise, the pairwise diagram for the tick species using the COI gene (Fig. 2d) demonstrated that D. laothaiensis was 87.7%, 88.8%, and 85.3% similar to D. steini from Thailand (ON680803), D. auratus from Thailand (ON680798) and a D. steini from Malaysia (MW971472). The total numbers of differentiation positions of nucleotide base sequences alignments were 46 (46 bases/ 0 gap), 42 (42 bases/ 0 gap), and 53 (53 bases/ 0 gap), respectively. While D. steini had 85.9% and 93.4% similarity compared with D. auratus from Thailand (ON680798) and D. steini from Malaysia (MW971472) and their total numbers of differentiation positions of nucleotide base sequences alignments were 53 (53 bases/ 0 gap) and 24 (24 bases/ 0 gap), respectively. Also, D. auratus (ON680798) had 98.7% similarity with D. auratus from Malaysia (MW971473), and the total numbers of differentiation positions of nucleotide base sequences alignments were 5 (5 bases/ 0 gap). All alignments of the related nucleotide bases and gap positions are shown in Fig. S2.

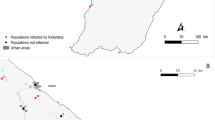
The location of the sampling site (black star) in Mae Sot district, Tak province, northwestern Thailand (neighboring Myanmar) (a). Indicated with a black dot, KP represents Khammouan province of Laos provided to enable a geographical comparison with Tak province of Thailand, where Ca. R. laoensis were detected. The numbers and species of Dermacentor tick with their SFGRs infection rates are provided. The photographic images of D. steini (b–d), D. laothaiensis (e–g), and D. auratus (h–j) tick morphologies are demonstrated with their different marked features. Figure 1 was designed and illustrated using PowerPoint 2019. (Photos: Aummarin Chaloemthanetphong; Map source: https://gistdaportal.gistda.or.th/portal/home/webmap/viewer.html?useExisting=1).

Phylogenetic analysis and the summary diagram of pairwise comparison among the tick species. Neighbor-joining trees based on partial gene sequences of the 16S rRNA (a) and COI (c) were constructed using the molecular evolutionary genetics analysis (MEGA) 7 software to represent three different species of the Dermacentor ticks from Tak province; D. laothaiensis (DLT), D. steini (DST), and D. auratus (DAT). The reference species for D. steini and D. auratus are included with their accession numbers, while D. laothaiensis shows the first molecular identification. The outgroup was D. reticulatus (MT478096). Only bootstrap values > 50% are shown above each branch. The pairwise diagrams correspond to the 16S rRNA (b) and COI (d) with their similarity percentages and the number of base/gap differences (b/g). The diagrams of pairwise differences were designed and illustrated using PowerPoint 2019.
Infection rate and molecular identification of SFGRs
The prevalence of rickettsiae from tick samples was investigated. The results using three housekeeping genes commonly used for SFGR screening, namely 17-kDa (434 bp), gltA (380 bp), and 16S rRNA (1,328 bp), all showed a positive rate of 6.10% (21/344). Further analyses of the amplified DNA samples employing three genes commonly used in Rickettsia genotyping, i.e., ompA (532 bp), ompB (1,295 bp), and sca4 (1,100 bp), clearly confirmed the presence of SFGRs in these positive ticks. The three species of ticks and their Rickettsia infection rates were as follows: 18.10% (19/105) in D. laothaiensis, 0.72% (1/139) in D. steini, and 1% (1/100) in D. auratus (Fig. 1). All positive amplicons were then sequenced and phylogenetic trees were constructed for the six individual targeted genes (17-kDa, gltA, 16S rRNA, ompA, ompB, and sca4). They showed more or less similar patterns of phylogenetic relationships with the closely related SFGRs from the NCBI database (Fig. 3). Our findings showed that there were two detected SFGRs in two separate clades as detailed below.


Phylogenetic analysis and the summary diagram of pairwise differences among Rickettsia species based on 17-kDa (a,b), gltA (c,d), 16S rRNA (e,f), ompA (g,h), ompB (i,j), and sca4 (k,l). The trees were constructed using the maximum-parsimony (MP) algorithms implemented in the molecular evolutionary genetics analysis (MEGA) 7 software. Bootstrap analysis was performed using the Subtree-Pruning-Regrafting (SPR) distances method (1,000 replications); only bootstrap values > 50% are shown. The black diamonds indicate the representative Rickettsia sequences of the samples in this study. The scale bar is in the units of substitution/site. The numbers in the summary diagram of pairwise differences among Ca. R. takensis, Ca. R. laoensis, and the references of related species of SFGRs indicate the percentages of nucleotide similarity and the number of base/gap/amino acid differences (b/g/a). The diagrams of pairwise differences were designed and illustrated using PowerPoint 2019.
Clade 1, the isolated clade, included only our novel genotype, Ca. R. takensis. Based on 17-kDa, the novel genotype showed phylogenetic relationships with R. japonica from China and Japan, Ca. R. vini from the Czech Republic, Rickettsia sp. TCM1 from Thailand and R. fournieri from Australia (Fig. 3a). However, its gltA was closely related to Rickettsia sp. ARRL2016-159 from Australia, R. japonica from China and Japan, and Ca. R. vini from the Czech Republic (Fig. 3c). The 16S rRNA gene, ompA, ompB, and sca4 all demonstrated their closest relationships with R. fournieri from Australia (Fig. 3e,g,i,k).
Clade 2 included another SFGR detected in the present study, Ca. R. laoensis (DAT10M3). This may be the first Thailand record of Ca. R. laoensis. Based on the 17-kDa gene, the Thailand specimen grouped with Ca. R. laoensis from Laos and Rickettsia sp. from India (Fig. 3a). Its gltA gene grouped with Rickettsia sp. from India, Rickettsia sp. RDla440 from the Thailand-Myanmar border and Ca. R. laoensis from Taiwan (Fig. 3c). On 16S rRNA, it grouped with Rickettsia sp. from Pakistan (Fig. 3e). Close phylogenetic relationships with Ca. R. laoensis were notably demonstrated for ompA from India and Laos (Fig. 3g), ompB and sca4 from Laos (Fig. 3i,k).
The results of these phylogenetic analyses supports the conclusion that there may be two unknown groups of Rickettsia spp. occurring in these tick vectors, which are tentatively referred to Ca. R. takensis and Ca. R. laoensis as mentioned above. However, some other SFGRs also showed phylogenetic relationships close to our discovered SFGRs. Therefore, to validate the species classification of our two putative detected SFGRs, their sequence alignments, and pairwise comparisons were performed against reference SFGRs. The results of the nucleotide similarity percentages determined were then applied to pose criteria for a novel Rickettsia determination using gene sequences20. The details of the nucleotide bases and amino acid variations are demonstrated in Figs. 3 and S3.
The highest percentage, indicating the closest similarity in each target gene is shown in bold font in the diagram. The 17-kDa gene which was used to screen Rickettsia in the population, showed the highest value (99.5%; 2 bases/ 0 gap/ 1 amino acid) for several SFGRs (R. fournieri from Australia, Ca. R. vini from the Czech Republic, and Rickettsia sp. TCM1 from Thailand; Fig. 3b). The gltA gene showed the highest value of similarity (99.7%, 1 base/ 0 gap/ 0 amino acid) for R. japonica from Thailand (PMK94), R. heilongjiangensis from Japan, and Rickettsia sp. ARRL2016-159 from Australia (Fig. 3d). For 16S rRNA the highest values of 99.4% (6 bases/ 1 gap) were obtained for R. fournieri from Australia, R. japonica from China (LA16/2015) and Japan (YHM and YM) (Fig. 3f). The ompA, ompB, and sca4 genes demonstrated similar results, finding that our Ca. R. takensis was closest to the R. fournieri from Australia. Their highest percentages and detail of variables were as follows: ompA 96.4% (12 bases/ 3 gaps/ 13 amino acids) (Fig. 3h), ompB 98.1% (15 bases/ 6 gaps/ 15 amino acids) (Fig. 3j), and sca4 98.9% (11 bases/ 0 gap/ 7 amino acids) (Fig. 3l). In addition, the pairwise comparisons between Ca. R. takensis and either Ca. R. laoensis (DAT10M3) from Thailand or Rickettsia sp. RDla440 from the Thailand-Myanmar border, showed exact similarity of 99.3% (2 bases/ 0 gap/ 1 amino acid) based on the only data available for the gltA gene (Fig. 3d). Based on the criteria for a novel Rickettsia determination using gene sequences of 16S rRNA, gltA, ompA, ompB, and sca4, an isolate can be classified as a new Rickettsia genotype when it exhibits no more than one of the following degrees of nucleotide similarity with the most homologous validated species (cutoff percentages ≥ 99.8, ≥ 99.9, ≥ 98.8, ≥ 99.2, and ≥ 99.3)20. The result of Ca. R. takensis fulfilled this criterion with the degrees of similarity 99.4, 99.7, 96.4, 98.1, and 98.9, respectively. None of the degrees were equal to or over the cutoff percentages of the standard criteria. This indicated the occurrence of a new Rickettsia genotype. Moreover, this Rickettsia was detected in ticks, not from rickettsial isolates: therefore Candidatus status could be applied. Therefore, this evidence showed that we found a novel genotype of SFGR in D. laothaiensis and D. steini tick samples. In addition, this novel Rickettsia was assigned as Candidatus Rickettsia takensis after the name of the collecting site, Tak province.
According to the evidence, we also found Ca. R. laoensis (DAT10M3) infection in one male in the D. auratus tick samples. The highest percentages of similarities based on the targeted genes were shown in pairwise comparisons. (1) DAT10M3 and Ca. R. laoensis from Laos ompA 100%, ompB 100%, and sca4 99.9% (1 base/ 0 gap/ 0 amino acid) (Fig. 3h, 3j, and 3l). (2) DAT10M3 and Ca. R. laoensis from China ompA 99.8% (1 base/ 0 gap / 0 amino acid) (Fig. 3h). (3) DAT10M3 and Rickettsia sp. from India 17-kDa 100%, gltA 100%, and ompA 100% (Fig. 3b,d,h). (4) DAT10M3 compared with either Rickettsia sp. from the Thailand-Myanmar border or Ca. R. laoensis from Taiwan gltA 100% (Fig. 3d). (5) DAT10M3 and Rickettsia sp. JC880 from Pakistan 16S rRNA 99.2% (8 bases/ 1 gap) (Fig. 3f). Overall, phylogenetic analyses and sequence alignments indicated that DAT10M3 and Ca. R. laoensis from Laos shared almost identical genotypes. This shows that Ca. R. laoensis is present on the northwestern border of Thailand. However, there have not yet been any reports on whether Ca. R. laoensis is a pathogen although it was detected in ticks (Haemaphysalis sp.16 and D. auratus in the present study).
Discussion
More than 40 species of Dermacentor ticks are known worldwide23,24,25. So far, eight species (D. auratus, D. compactus, D. steini, D. filippovae, D. pasteuri, D. laothaiensis, D. falsosteini, and D. tricuspis) have been found in Thailand24,25,26,27,28. Of these, three species were morphologically and molecularly identified as D. laothaiensis, D. steini, and D. auratus in the present study. D. laothaiensis was recently described, and it was reported as morphologically closely related to D. steini26. However, we found that D. laothaiensis was quite distinct from D. steini as these two species resolving into separate clades during phylogenetic analyses. The distinction was further supported as data was harvested from nucleotide sequence alignments (Fig. S2) demonstrating differentiation during pairwise comparisons among the tick species (Fig. 2). The results of our molecular analyses and phylogenetic relationships based on 16S rRNA and COI genes constitute the first molecular evidence indicating the difference between the D. laothaiensis and D. steini, and also showed that both were distinct from D. auratus.
In Thailand, several studies have examined the detection of Rickettsia in Dermacentor ticks collected from vegetation and mammal hosts. Malaisri et al. obtained 13 adult-stage Dermacentor ticks from vegetation and observed no evidence of rickettsial infection29. Sumrandee et al. collected a total of 16 Dermacentor ticks (15 adults and 1 nymph) from Artiodactyla and identified rickettsial infection in 5 of them, accounting for 31.3% (5/16)30. Nooroong et al. (2018) collected 114 adult Dermacentor ticks from vegetation, of which 5 were infected with Rickettsia, representing 4.4% (5/114)31. Takhampunya et al. investigated 58 Dermacentor ticks, consisting of 35 larvae and 23 adults, and detected rickettsial infection in only 1 adult, accounting for 1.7% (1/58)32. Finally, Hirunkanokpun et al. examined 8 Dermacentor auratus nymphs from a Burmese ferret badger but found no rickettsial infection in these nymphs18. These studies indicate a rare or relatively low incidence of rickettsial infection in Dermacentor ticks, except for the findings of Sumrandee et al. with a rate of 31.3%30.
Reviews of SFGRs in Thailand1 have previously revealed at least 5 defined species (R. honei, R. japonica, R. helvetica, R. rickettsii, and R. thailandii). In addition, another 4 SFGRs were also reported as genotypically related to R. raoultii, R. tamurae, R. monacensis, and R. montana1,29,30,31,32,33,34. Globally distributed SFGRs comprise more than 20 species3of which over 40% are likely found in Thailand. However, only a few studies have been conducted there compared to other regions. Interestingly, several SFGRs were reported from the Thailand-Myanmar border. In 1994, SFGR antibodies for R. japonica and R. honei (TT-118) were detected in rat serum collected in the 1970s at Kanchanaburi6 (14°N 99°E). In 2003, Rickettsia sp. RDa420 infecting D. auratus and Rickettsia sp. RDla440 infecting Dermacentor larvae were also reported from Kanchanaburi12. In the present study, there were two different genotypes of SFGRs, one of which was proposed as a novel genotype, Ca. R. takensis, and the other provided Thailand’s first record of Ca. R. laoensis in Dermacentor ticks. Both were collected from our study area in Tak (16°N 99°E), another border province with the same longitude comparing with Kanchanaburi province as mentioned above that there were some reports of SFGRs. This may indicate the distribution of SFGRs along the Thailand-Myanmar border.
In this study, our phylogenetic classification of Ca. R. takensis lacked clarity because its genotype showed close relationships to (1) R. fournieri from Australia and R. japonica from China and Japan (16S rRNA) (2) Rickettsia sp. from Australia, R. japonica from Thailand, and R. heilongjiangensis from Japan (gltA) and (3) R. fournieri from Australia (ompA, ompB, and sca4) (Fig. 3d,f,h,j,l). However, individually targeted gene trees demonstrated a unique Rickettsia genotype resolving separately from the other reference clades (Fig. 3a,c,e,g,i,k). This was likely supported by the results summarized in the diagrams of Fig. 3. The criteria for classifying Rickettsia species20 were applied, and it was confirmed that Ca. R. takensis was a member of the genus Rickettsia according to its 16S rRNA and gltA homology against some other validated Rickettsia spp. by the degree of homology of ≥ 98.1% and ≥ 86.5%, respectively for these two genes. Furthermore, the presence of the ompA gene also confirmed that it belonged to the SFGRs. Again, when we examined Rickettsia species identification by MLST, we found that in no case was the degree of nucleotide similarity (99.4, 99.7, 96.4, 98.1, and 98.9), compared to the most homologous species from the NCBI database (Fig. 3 and Table 1), equal to or over the respective standard criteria cutoff percentages (≥ 99.8, ≥ 99.9, ≥ 98.8, ≥ 99.2, and ≥ 99.3). Therefore, these results indicated the uniqueness of a novel genotype of Ca. R. takensis reported in this study. Moreover, current investigations of rickettsiae associated with Dermacentor ticks in the vicinity of the Thailand-Myanmar border showed the presence of a novel genotype, Ca. R. takensis, predominantly detected in D. laothaiensis and to a lesser extent in D. steini. Ca. R. takensis found in all 20 ticks exhibited similar nucleotide sequences for the six genes (17-kDa, gltA, 16S rRNA, ompA, ompB, and sca4) examined. Surprisingly, only one male of D. auratus was infected with Ca. R. laoensis (DAT10M3). Pairwise comparisons of nucleotide sequences of these two genotypes of rickettsiae based on the targeted genes showed 97.4%, 99.3%, 98.6%, 89.9%, 95.2%, and 97.5% similarities, respectively. These data confirmed that our detected SFGRs, either Ca. R. takensis or Ca. R. laoensis, were two distinct Rickettsia species that infected the population of our tick samples.
Another differentiated SFGRs genotype, Ca. R. laoensis (DAT10M3), was classified and this yielded the first molecular evidence for its presence in Thailand. Importantly, based on the gltA gene, our sample DAT10M3 exhibited a close relationship with the Rickettsia sp. RDla440, the SFGRs reported in Dermacentor larvae from Kanchanaburi12. However, the gltA gene sequence analysis was the only available data for RDla440, although it yielded 100% similarity compared to our DAT10M3 (299 bp) sequence alignment. Subsequently it was reported that RDla440 appeared to be more closely related to the sequences of Rickettsia sp. strain DnS14 and Rickettsia sp. strain RpA4, differing in only 2 bp (99.7% similarity) based on gltA (1,108 bp)12. However, both DnS14 and RpA4 were later identified as R. raoultii based on 16S rRNA, gltA, ompA, ompB, and sca4 genes35. Therefore, although the results with the gltA gene indicated DAT10M3 had 99.7% similarity (variable of 1 base) with either DnS14 or RpA4 strains detected in ticks from the former Soviet Union36, data from a single gene might be insufficient for SFGRs classification. Likewise, based on our results, our sample DAT10M3 was most likely Ca. R. laoensis, related to samples of Ca. R. laoensis reported in Laos16. However, it was not certain whether the RDla440 detected earlier at the same border longitude of this was in fact Ca. R. laoensis due to the limited molecular evidence. Although gltA has frequently been used as a target for generic diagnostics based on PCR because this approach can easily identify a number of Rickettsia37,38,39, this gene is also highly conserved. Therefore, gltA on its own might not be enough to differentiate between closely related Rickettsia species. Consequently, the MLST of 17-kDa, gltA, 16S rRNA, ompA, ompB, and sca4 were suggested as potentially helpful and appropriate for the reconstruction of the evolutionary relationship of diverse but closely related Rickettsia species, as documented in our results and previous reports1,2,16,38,40,41.
Ca. R. laoensis infecting Haemaphysalis nymphs (4.5%) was first reported in Khammouan province, Laos16. We found that Ca. R. laoensis in Thailand and Laos showed the highest percentage values during pairwise comparison of the five genes, 17-kDa, gltA, ompA, ompB, and sca4, with 99.5%, 99.6%, 100%, 100%, and 99.9% similarity, respectively. Khammouan province in Laos is located near the northeastern border of Thailand, bordering the Mekong River, about 700 km east of Tak province which lies on the Myanmar border (Fig. 1). This geographical evidence seems to suggest that Ca. R. laoensis is probably a common species, and widely distributed in Thailand, at least in the upper part of the country. In addition, Wang et al. (2020) found Ca. R. laoensis infecting 4 species of Haemaphysalis (H. bispinosa, H. longicornis, H. flava, and H. hystricis) and 4 species of Dermacentor (D. auratus, D. atrosignatus, D. silvarum, and D. taiwanensis) in eastern China, based on the nucleotide sequence of ompA gene (MT321615)42. These authors also showed that Ca. R. laoensis in China was molecularly similar to the Rickettsia sp. (MK905251) infecting Haemaphysalis ticks in the Western Ghats, India (Arunkumar, G., GenBank in NCBI, 2019). Although, the nucleotide sequences of 3 genes (17-kDa, gltA, and ompA) showed that Ca. R. laoensis from Thailand and India have close relationship (Table 1). And recently, Yen et al. (2022) also reported Ca. R. laoensis infecting D. auratus in Taiwan, based on nucleotide sequences of four genes; gltA (MZ869826), ompA (MZ869827), ompB (MZ869829), and sca4 (MZ869830)41. Taken together, these data suggest that Ca. R. laoensis from Thailand, Laos, China, Taiwan, and India may represent a complex of species within this Rickettsia cluster. This complex cluster reflects species diversity of SFGRs in Asia, centered around Southeast Asia, including Thailand. As well as seeking to detect Ca. R. laoensis in different species of tick vectors in this region, more information about its pathogenicity is needed. Therefore, more fieldwork investigating Ca. R. laoensis infection in ticks and humans in this region is required. Although the information on species diversity of rickettsiae related to the emerging rickettsioses via tick vectors is still poorly understood in Thailand, our study has enhanced knowledge of SFGRs harbored in these 3 species of Dermacentor ticks in this particular area of northwestern Thailand. Thus, the prevalence of these SFGRs infected tick vectors along the border of Thailand and Myanmar, extending for approximately 2,400 km from the north to the south, will be very useful and informative because these areas are gateways for the movement of local people between the two countries. The border areas also involve legal and illegal livestock trading, increasing the potential risk of SFGRs exposure in this region.
Furthermore, the polymorphic genotypes of Ca. R. takensis and Ca. R. laoensis discovered in this study indicated the genetic diversity of SFGRs in this area. In addition, understanding the movements of wildlife, especially mammalian hosts of ticks, may contribute to the knowledge of the distribution of rickettsial agents in this particular forested area. The data seem to suggest a potential risk for the distribution of this SFGRs in neighboring countries, including Thailand, Laos, and Myanmar. The diagnosis and management of rickettsial infections in this region remains challenging, for instance, the results of clinical differential diagnoses for SFGRs are usually disregarded in resource-poor settings because of the limitations of immunofluorescence serologic tests caused by antigenic cross-reactions43. While the identification of phylogenetically distinct SFGRs to species by molecular methods is well known to have practical applications, public health facilities and attention related to rickettsiae needs to be improved to enable greater accessibility in Southeast Asia.
Materials and methods
Tick collection and species identification
A total of 344 adults of Dermacentor ticks were collected from vegetation in the forest of a tourist trail (with shrubs and trees along both sides) at Taksin Maharat National Park, Mae Sot district, Tak province, about 20 km from the Thailand-Myanmar border (Fig. 1a). The collecting of tick samples was conducted upon approval from the Department of National Parks, Wildlife and Plant Conservation of Thailand (permission no. TS 0907.4/10,680). Collections were made at 754 m asl, and ticks were searched for and collected, with blunt forceps or hand picking from vegetation in 2016 with an average temperature of 23 °C, and an average humidity of 80% for the environmental conditions. All tick samples were kept in 70% ethanol and stored at −20 °C for laboratory study. Each tick was morphologically identified using available descriptions26,44,45. The samples of these ticks were subsequently confirmed by molecular techniques using DNA sequencing of two genes; 16S ribosomal RNA (16S rRNA: 16S + 1, 16S-1) and cytochrome c oxidase I (COI: RON, TCOIR), as described by Black and Piesman and Simon et al.46,47. The references for primers used are listed in Table 2.
DNA extraction
DNA extractions of all tick samples were performed after they had been morphologically identified. An individual tick was serially rinsed and cleaned thoroughly in 70% ethanol, 10% sodium hypochlorite (NaOCl), and sterilized distilled water three times (1 min each) to avoid external bacterial contamination. The total DNA of each tick sample was extracted from the whole body of individual adults using the QIAamp DNA Extraction Mini Kit for Tissue (QIAGEN, Germany) according to the manufacturer's instructions. Then, 16S rRNA amplification was used to monitor the DNA extraction quality before the product was stored at − 20 °C for further study.
Rickettsia detection, identification and sequencing analyses
PCR was performed to amplify rickettsial DNA from extracted products of each Dermacentor tick. Overall, six targeted partial fragments of genes were used (Table 2). Initially, detection and screening to determine the bacteria of the genus Rickettsia were carried out employing the genus-common 17-kDa surface antigen gene (17-kDa), the pan bacterial gene encoding 16S rRNA (16S rRNA) and the citrate synthase gene (gltA). Rickettsia-positive rates were then calculated. Additionally, we selected positive samples to test with other PCR amplifications that targeted the SFGRs-specific 190-kDa outer membrane protein A gene (ompA), the 120-kDa outer membrane protein B gene (ompB), and the PS120 or D protein-encoding gene (sca4) to determine their SFGRs features. The PCR amplicons of 17-kDa 434 bp, 16S rRNA 1,328 bp, ompA 532 bp, ompB 1,295 bp, and sca4 1,100 bp were purified using the NucleoSpin Gel and PCR Clean‑up Mini kit (MACHEREY–NAGEL, Germany). The purified amplicons were sequenced in both directions at Macrogen Co., LTD (Seoul, Korea). Furthermore, any ambiguous positive PCR products were gel purified, inserted into the pGEM® -T easy vector system (Promega, USA), and transformed into E. coli (strain JM109) using standard cloning protocols (or according to the manufacturer’s protocol). A positive cloning colony was grown on an LB-agar plate (Luria–Bertani) containing 20 mg/ml IPTG, 20 mg/ml X-Gal and 100 μg/ml ampicillin. The recombinant plasmids were also extracted and subjected to DNA sequencing.
Data analysis
Data analyses of the obtained nucleotide sequences were primarily performed to determine the highest similarity using the Basic Local Alignment Search Tool (BLAST)52. Then, multiple sequence alignments with the related DNA sequences (Table S1 and S2) retrieved from the GenBank database were aligned by the CLUSTAL method, BioEdit v.2.0.0 program, while the amino acid polymorphisms were determined using the Amino Acid Explorer of the NCBI server52.
Phylogenetic trees were constructed using Neighbor-joining (NJ) method based on 16S rRNA and COI genes for tick species identification and Maximum-parsimony (MP) method based on 17-kDa, gltA, 16S rRNA, ompA, ompB, and sca4 genes for rickettsial genotype identification. These analyses were performed in the Molecular Evolutionary Genetics Analysis (MEGA) 7.0 software53. Distance matrix analysis was generated by the Tamura 3-parameters with gamma distribution (T92 + G) and Subtree-Pruning-Regrafting (SPR) for multiple substitutions. Bootstrap values were based on 1,000 replicates to estimate the support for nodes within the phylogenetic trees.
Ethics approval
This research was approved by the SCMU-ACUC committee, protocol No. MUSC64-012-561.
Data availability
The nucleotide sequences of the Rickettsia obtained in this study are deposited to GenBank database (https://www.ncbi.nlm.nih.gov/genbank/).
References
Low, V. L., Tan, T. K., Khoo, J. J., Lim, F. S. & AbuBakar, S. An overview of rickettsiae in Southeast Asia: Vector-animal-human interface. Acta Trop. 202, 105282. https://doi.org/10.1016/j.actatropica.2019.105282 (2020).
Fournier, P. E. & Raoult, D. Current knowledge on phylogeny and taxonomy of Rickettsia spp.. Ann. N. Y. Acad. Sci. 1166, 1–11. https://doi.org/10.1111/j.1749-6632.2009.04528.x (2009).
Parola, P. et al. Update on tick-borne rickettsioses around the world: A geographic approach. Clin. Microbiol. Rev. 26, 657–702. https://doi.org/10.1128/CMR.00032-13 (2013).
Merhej, V., Angelakis, E., Socolovschi, C. & Raoult, D. Genotyping, evolution and epidemiological findings of Rickettsia species. Infect. Genet. Evol. 25, 122–137. https://doi.org/10.1016/j.meegid.2014.03.014 (2014).
Satjanadumrong, J., Robinson, M. T., Hughes, T. & Blacksell, S. D. Distribution and ecological drivers of spotted fever group Rickettsia in Asia. EcoHealth 16, 611–626. https://doi.org/10.1007/s10393-019-01409-3 (2019).
Parola, P. et al. Emerging rickettsioses of the Thai-Myanmar border. Emerg. Infect. Dis. 9, 592–595. https://doi.org/10.3201/eid0905.020511 (2003).
Pickard, A. L. et al. A study of febrile illnesses on the Thai-Myanmar border: Predictive factors of rickettsioses. Southeast Asian J. Trop. Med. Public Health. 35, 657–663 (2004).
Fournier, P. E. et al. Aneruptive fever associated with antibodies to Rickettsia helvetica in Europe and Thailand. J. Clin. Microbiol. 42, 816–818. https://doi.org/10.1128/JCM.42.2.816-818.2004 (2004).
Jiang, J. et al. Human infection with Rickettsia honei Thailand. Emerg. Infect. Dis. 11, 1473–1475. https://doi.org/10.3201/eid1109.050011 (2005).
Sangkasuwan, V. et al. Description of the first reported human case of spotted fever group rickettsiosis in urban Bangkok. Am. J. Trop. Med. Hyg. 77, 891–892. https://doi.org/10.4269/ajtmh.2007.77.891 (2007).
Gaywee, J. et al. Human infection with Rickettsia sp. related to R. japonica Thailand. Emerg. Infect. Dis. 13, 657. https://doi.org/10.3201/eid1304.060585 (2007).
Parola, P. et al. Detection of Ehrlichia spp., Anaplasma spp., Rickettsia spp., and other eubacteria in ticks from the Thai-Myanmar border and Vietnam. J. Clin. Microbiol. 41, 1600–1608. https://doi.org/10.1128/JCM.41.4.1600-1608.2003 (2003).
Takada, N. et al. Spotted fever group Rickettsia sp. closely related to R. japonica Thailand. Emerg. Infect. Dis. 15, 610. https://doi.org/10.3201/eid1504.071271 (2009).
Kollars, T. M., Tippayachai, B. & Bodhidatta, D. Thai tick typhus, Rickettsia honei, and a unique rickettsia detected in Ixodes granulatus (Ixodidae: Acari) from Thailand. Am. J. Trop. Med. 65, 535–537. https://doi.org/10.4269/ajtmh.2001.65.535 (2001).
Sumrandee, C. et al. Molecular detection of Rickettsia species in Amblyomma ticks collected from snakes in Thailand. Ticks Tick Borne Dis. 5, 632–640. https://doi.org/10.1016/j.ttbdis.2014.04.013 (2014).
Taylor, A. J. et al. Large-scale survey for tickborne bacteria, Khammouan Province. Laos. Emerg. Infect. Dis. 22, 1635. https://doi.org/10.3201/eid2209.151969 (2016).
Kho, K. L., Tan, P. E. & Tay, S. T. Diversity of rickettsiae in feeding and questing ticks collected from a Malaysian forest reserve area. J. Med. Entomol. 56, 547–552. https://doi.org/10.1093/jme/tjy168 (2019).
Hirunkanokpun, S., Ahantarig, A., Baimai, V., Pramual, P. & Trinachartvanit, W. A new record of Rickettsia japonica in ticks infesting a Burmese ferret-badger in Thailand. Trop. Biomed. 39, 55–59. https://doi.org/10.47665/tb.39.1.007 (2022).
Niebylski, M. L. et al. Rickettsia peacockii sp. nov, a new species infecting wood ticks, Dermacentor andersoni, in western Montana. Int. J. Syst. Bacteriol. 47, 446–452. https://doi.org/10.1099/00207713-47-2-446 (1997).
Fournier, P. E. et al. Gene sequence-based criteria for identification of new Rickettsia isolates and description of Rickettsia heilongjiangensis sp. Nov.. J. Clin. Microbiol. 41, 5456–5465. https://doi.org/10.1128/JCM.41.12.5456-5465.2003 (2003).
Diop, A. et al. Rickettsia fournieri sp. Nov., a novel spotted fever group rickettsia from Argas lagenoplastis ticks in Australia. Int. J. Syst. Evol. Microbiol. 68, 3781–3784. https://doi.org/10.1099/ijsem.0.003057 (2018).
Li, H. et al. Candidatus Rickettsia xinyangensis as cause of spotted fever group rickettsiosis, Xinyang, China, 2015. Emerg. Infect. Dis. 26, 985–988. https://doi.org/10.3201/eid2605.170294 (2020).
Guglielmone, A. A., Petney, T. N. & Robbins, R. G. Ixodidae (Acari: Ixodoidea): Descriptions and redescriptions of all known species from 1758 to December 31, 2019. Zootaxa 4871, 1–322. https://doi.org/10.11646/zootaxa.4871.1.1 (2020).
Guglielmone, A. A. et al. Individual species accounts in The Hard Ticks of the World: (Acari: Ixodida: Ixodidae) (eds. Guglielmone, A. A. et al.) 609–645 (Springer, 2014).
Apanaskevich, D. A. & Apanaskevich, M. A. Description of a new Dermacentor (Acari: Ixodidae) species from Thailand and Vietnam. J. Med. Entomol. 52, 806–812. https://doi.org/10.1093/jme/tjv067 (2015).
Apanaskevich, D. A. et al. Description of a new species of Dermacentor Koch, 1844 (Acari: Ixodidae) from Laos and Thailand. Syst. Parasitol. 96, 475–484. https://doi.org/10.1007/s11230-019-09861-z (2019).
Apanaskevich, D. A. et al. Description of a new species of Dermacentor Koch, 1844 (Acari: Ixodidae) from the mountains of Laos and Thailand. Syst. Parasitol. 97, 347–355. https://doi.org/10.1007/s11230-020-09916-6 (2020).
Apanaskevich, D. A., Apanaskevich, M. A., Nooma, W., Ahantarig, A. & Trinachartvanit, W. Reinstatement of Dermacentor tricuspis (Schulze, 1933) n. comb., n. stat. (Acari: Ixodidae) as a valid species, synonymization of D.atrosignatus Neumann, 1906 and description of a new species from Indonesia Malaysia and Thailand. Syst. Parasitol. 98, 207–230. https://doi.org/10.1007/s11230-021-09972-6 (2021).
Malaisri, P., Hirunkanokpun, S., Baimai, V., Trinachartvanit, W. & Ahantarig, A. Detection of Rickettsia and Anaplasma from hard ticks in Thailand. J. Vector Ecol. 40, 262–268. https://doi.org/10.1111/jvec.12163 (2015).
Sumrandee, C., Baimai, V., Trinachartvanit, W. & Ahantarig, A. Molecular detection of Rickettsia, Anaplasma, Coxiella and Francisella bacteria in ticks collected from Artiodactyla in Thailand. Ticks Tick Borne Dis. 7, 678–689. https://doi.org/10.1016/j.ttbdis.2016.02.015 (2016).
Nooroong, P., Trinachartvanit, W., Baimai, V. & Ahantarig, A. Phylogenetic studies of bacteria (Rickettsia, Coxiella, and Anaplasma) in Amblyomma and Dermacentor ticks in Thailand and their co-infection. Ticks Tick Borne Dis. 9, 963–971. https://doi.org/10.1016/j.ttbdis.2018.03.027 (2018).
Takhampunya, R. et al. The bacterial community in questing ticks from Khao Yai National Park in Thailand. Front. Vet. Sci. 8, 1335. https://doi.org/10.3389/fvets.2021.764763 (2021).
Hirunkanokpun, S., Kittayapong, P., Cornet, J. P. & Gonzalez, J. P. Molecular evidence for novel tick-associated spotted fever group rickettsiae from Thailand. J. Med. Entomol. 40, 230–237. https://doi.org/10.1603/0022-2585-40.2.230 (2003).
Doornbos, K. et al. Rickettsia sp. closely related to Rickettsia raoultii (Rickettsiales: Rickettsiaceae) in an Amblyomma helvolum (Acarina: Ixodidae) tick from a Varanus salvator (Squamata: Varanidae) in Thailand. J. Med. Entomol. 50, 217–220. https://doi.org/10.1603/ME12010 (2013).
Mediannikov, O. et al. Rickettsia raoultii sp. Nov., a spotted fever group rickettsia associated with Dermacentor ticks in Europe and Russia. Int. J. Syst. Evol. Microbiol. 58, 1635–1639. https://doi.org/10.1099/ijs.0.64952-0 (2008).
Rydkina, E. et al. New rickettsiae in ticks collected in territories of the former soviet union. Emerg. Infect. Dis. 5, 811–814. https://doi.org/10.3201/eid0506.990612 (1999).
Roux, V., Rydkina, E., Eremeeva, M. & Raoult, D. Citrate synthase gene comparison, a new tool for phylogenetic analysis, and its application for the rickettsiae. Int. J. Syst. Bacteriol. 47, 252–261. https://doi.org/10.1099/00207713-47-2-252 (1997).
Jiang, J., Stromdahl, E. Y. & Richards, A. L. Detection of Rickettsia parkeri and Candidatus Rickettsia andeanae in Amblyomma maculatum gulf coast ticks collected from humans in the United States. Vector Borne Zoonot. Dis. 12, 175–182. https://doi.org/10.1089/vbz.2011.0614 (2012).
Anderson, B. E. & Tzianabos, T. Comparative sequence analysis of a genus common rickettsial antigen gene. J. Bacteriol. Res. 171, 5199–5201. https://doi.org/10.1128/jb.171.9.5199-5201.1989 (1989).
Loyola, S. et al. Molecular characterization by multilocus sequence typing and diversity analysis of Rickettsia asembonensis in Peru. Vector Borne Zoonot. Dis. 22, 170–177. https://doi.org/10.1089/vbz.2021.0077 (2022).
Yen, T. Y. et al. Seroepidemiological study of spotted fever group rickettsiae and identification of a putative new species, Rickesttsia sp. Da-1, in Gongliao, Northeast Taiwan. Pathogens 10, 1434. https://doi.org/10.3390/pathogens10111434 (2021).
Wang, X. et al. Identification and molecular analysis of Ixodid ticks (Acari: Ixodidae) infesting wild boars (Sus scrofa) and tick-borne pathogens at the Meihua mountain of southwestern Fujian, China. Vet. Parasitol. 22, 100492. https://doi.org/10.1016/j.vprsr.2020.100492 (2020).
Aung, A. K., Spelman, D. W., Murray, R. J. & Graves, S. Rickettsial infections in Southeast Asia: Implications for local populace and febrile returned travelers. Am. J. Trop. Med. Hyg. 91, 451. https://doi.org/10.4269/ajtmh.14-0191 (2014).
Wassef, H. Y. & Hoogstraal, H. Dermacentor (Indocentor) auratus (Acari: Ixodoidea: Ixodidae): Identity of male and female. J. Med. Entomol. 21, 169–173. https://doi.org/10.1093/jmedent/21.2.169 (1984).
Wassef, H. Y. & Hoogstraal, H. Dermacentor (Indocentor) steini (Acari: Ixodoidea: Ixodidae): Identity of male and female. J. Med. Entomol. 23, 532–537. https://doi.org/10.1093/jmedent/23.5.532 (1986).
Black, W. C. & Piesman, J. Phylogeny of hard-and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc. Natl. Acad. Sci. U. S. A. 91, 10034–10038. https://doi.org/10.1073/pnas.91.21.10034 (1994).
Simon, C. et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701. https://doi.org/10.1093/aesa/87.6.651 (1994).
Webb, L., Carl, M., Malloy, D. C., Dasch, G. A. & Azad, A. F. Detection of murine typhus infection in fleas by using the polymerase chain reaction. J. Clin. Microbiol. Infect. 28, 530–534. https://doi.org/10.1128/jcm.28.3.530-534.1990 (1990).
Anstead, C. A. & Chilton, N. B. A novel Rickettsia species detected in vole ticks (Ixodes angustus) from Western Canada. Appl. Environ. Microbiol. 79, 7583–7589. https://doi.org/10.1128/AEM.02286-13 (2013).
Regnery, R. L., Spruill, C. L. & Plikaytis, B. D. Genotypic identification of rickettsiae and estimation of intraspecies sequence divergence for portions of two rickettsial genes. J. Bacteriol. 173, 1576–1589. https://doi.org/10.1128/jb.173.5.1576-1589.1991 (1991).
Roux, V. & Raoult, D. Phylogenetic analysis of members of the genus Rickettsia using the gene encoding the outer-membrane protein rOmpB (ompB). Int. J. Syst. Evol. Microbiol. 50, 1449–1455. https://doi.org/10.1099/00207713-50-4-1449 (2000).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Acknowledgements
We thank the Department of National Parks, Wildlife and Plant Conservation of Thailand (Taksin Maharat National Park, approval number TS0907.4/10680) for field work cooperation. We also thank Associate Professor Philip D. Round and Dr. Adrian R. Plant for invaluable comments. This research was partially supported by Mahidol University and the Center of Excellence on Biodiversity, Office of Higher Education Commission (BDC-PG3-161006).
Author information
Authors and Affiliations
Contributions
A.C. carried out the field collection of samples, performed molecular experiments, analyzed data and drafted the article. W.T., V.B., and A.A. contributed in planning the conception and experimental design. W.T. in charge of overall direction and planning with guidance from V.B. D.A.A. supervised tick collection and taxonomy. A.A. and S.H. supervised molecular experiments and analyses. All authors discussed the results, revised the manuscript, read and approved the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaloemthanetphong, A., Ahantarig, A., Apanaskevich, D.A. et al. A novel Rickettsia, Candidatus Rickettsia takensis, and the first record of Candidatus Rickettsia laoensis in Dermacentor from Northwestern Thailand. Sci Rep 13, 10044 (2023). https://doi.org/10.1038/s41598-023-37206-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-37206-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
