
Abstract
Intellectual disability (ID), which presents itself during childhood, belongs to a group of neurodevelopmental disorders (NDDs) that are clinically widely heterogeneous and highly heritable, often being caused by single gene defects. Indeed, NDDs can be attributed to mutations at over 1000 loci, and all type of mutations, ranging from single nucleotide variations (SNVs) to large, complex copy number variations (CNVs), have been reported in patients with ID and other related NDDs. In this study, we recruited seven different recessive NDD families with comorbidities to perform a detailed clinical characterization and a complete genomic analysis that consisted of a combination of high throughput SNP-based genotyping and whole-genome sequencing (WGS). Different disease-associated loci and pathogenic gene mutations were identified in each family, including known (n = 4) and novel (n = 2) mutations in known genes (NAGLU, SLC5A2, POLR3B, VPS13A, SYN1, SPG11), and the identification of a novel disease gene (n = 1; NSL1). Functional analyses were additionally performed in a gene associated with autism-like symptoms and epileptic seizures for further proof of pathogenicity. Lastly, detailed genotype-phenotype correlations were carried out to assist with the diagnosis of prospective families and to determine genomic variation with clinical relevance. We concluded that the combination of linkage analyses and WGS to search for disease genes still remains a fruitful strategy for complex diseases with a variety of mutated genes and heterogeneous phenotypic manifestations, allowing for the identification of novel mutations, genes, and phenotypes, and leading to improvements in both diagnostic strategies and functional characterization of disease mechanisms.
Similar content being viewed by others


Introduction
Intellectual disability (ID) is a neurodevelopmental disorder (NDD) characterized by significantly impaired intellectual and adaptive function. ID affects about 2–3% of the general population, with the majority of the cases (75–90%) suffering from mild ID and with only 10–25% of the cases reporting moderate or severe ID1. It is subdivided into two subtypes, known as non-syndromic ID (NSID), in which cognitive impairment is the only apparent clinical symptom, as further phenotyping (MRI, biochemical profiling, and so on) often reveals additional symptoms, and syndromic ID (SID), in which intellectual deficits are accompanied by other neurological and behavioral manifestations. ID is also frequently seen in autistic children, who exhibit deficits in social communication and interaction, repetitive behaviors, and/or restricted interests2,3,4.
The majority of ID disorders are caused by single genetic defects5, the discovery of which is vastly accelerating due to advances in genomics and high throughput sequencing. High throughput sequencing in Autism Spectrum Disorders (ASD) has led to the identification of a large number of rare variations in both DNA sequence and chromosomal structure conferring disease risk6,7. Given the large number of genes and genetic variations implicated in NDD syndromes, a thorough screening of the genome is the most suitable molecular technique to identify the disease-causing genomic events.
In this study, we examined seven different families featuring complex NDD syndromes, in which ID was accompanied by other neurodegenerative and/or metabolic manifestations, and performed homozygosity mapping (HM) through high throughput SNP genotyping and WGS to identify the disease-associated loci and pathogenic and genetic variations. All but one of the examined pedigrees belong to consanguineous recessive marriages, in which the affected subjects, due to identity by descent, are likely to have two identical copies of the disease allele derived from a common ancestor; thus, reducing genetic heterogeneity. Herein, the use of HM to discover disease-associated loci and reduce variant screening in subsequent WGS analyses is particularly powerful in these families. We identified pathogenic variations in seven different disease genes and carried out functional assays in one disease gene associated with both ID and autistic-like symptoms. Detailed clinical characterization was also performed in family with the objective to assist with the genotype-phenotype correlations and clinical diagnoses of prospective families.
Results
Clinical findings
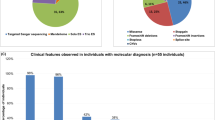
We recruited and clinically characterized seven different families featuring complex NDD disorders (Fig. 1A). The detailed clinical descriptions for each individual family are compiled in Table 1. Briefly, five families presented with ID and movement disorder phenotypes, including ataxia, spastic paraplegia, and chorea-acanthocytosis, among others, and two families presented with severe ID that was accompanied with autistic-like features as well as language behaviors in one family. Autistic, social and psychiatric behaviors were also observed in one family with ID and motor abnormalities; and epileptic seizures, which were treated with broad spectrum and different anti-epileptic drugs, including levetiracetam, carmazepin, and phenobarbital, were reported in four different families (Fig. 1, Table 1). Although most families presented with different phenotypic manifestations, all presented ID, and most of them featured some kind of motor abnormality.

Pedigree structures of families presenting with ID syndromes and their corresponding pathogenic mutations. The pedigrees of seven families featuring NDD syndromes are shown. Affected members are represented by either dark squares (males) or circles (females). The only individual who is non-manifesting for the phenotype but is homozygous for the mutation is represented with a white square with a black dot in the middle. Individuals with homozygous mutations are represented as m/m; heterozygous carriers as wt/m; and non-carriers individuals homozygous for the healthy allele as wt/wt. *Indicates those individuals that were subject to WGS analyses. The Sanger chromatogram sequences of the identified pathogenic mutations highlighting the homozygous mutant alleles (red arrow) are shown below each pedigree.
Genetic findings
Because the performed HM analyses identified several disease-associated loci in each individual family (Table 2), WGS analyses (Fig. 2) were then carried out in one (Family 7) or two (the remaining families) affected members from each individual family. These analyses led to the identification of several rare coding variations (Table 2), but only rare genetic variations within the associated loci were considered further (Fig. 2). Six out of seven families were found to carry pathogenic mutations in known NDD genes, with four mutations already reported and known to be associated with clinical phenotypes similar to those observed in our recruited families; while the two other, located in SLC5A2 (solute carrier family 5 member 2) and SYN1 (synapsin 1) genes, were novel and found to be absent in ethnicity-matched neurological individuals and public databases (see WGS methods for more details). Subsequent Sanger sequencing analyses revealed that all identified mutations segregated with the disease status.

Flow chart for WGS analysis. It shows the steps carried out in the recruited DNA samples for accurate and reliable disease-gene identification.
The affected subjects from Family Fam-01, who presented with severe ID and epilepsy but without behavioral abnormalities, were found to carry a known pathogenic mutation (p.Asp312Asn) in the NAGLU (N-acetyl-alpha-glucosaminidase) gene. This mutation was previously reported in patients with Sanfilippo syndrome B or mucopolysaccharidosis type III (MPS III) – a lysosomal storage disease characterized by behavioral changes including hyperactivity, aggressiveness, and destructive behaviors that progress to profound cognitive impairment and severe disability8 – and in a patient with severe ID9. Patients from family Fam-02, who presented with ID and motor abnormalities, were found to carry a novel mutation (p.Cys361Tyr) in the SLC5A2 gene, also known as sodium-glucose transporter 2 (SGLT2). Mutations in SLC5A2 are known to cause renal glycosuria10, which was also a symptom found in our patients with SLC5A2 mutations. This mutation was absent in 182 ethnicity-matched control chromosomes and public databases and was predicted to be pathogenic by all the computational methods used, including Mutation Taster (disease-causing), MutPred (0.842), SIFT (damage), SNPs&Go (disease-causing; 0.837), and CADD (25.5). Patients from family Fam-03 manifested a complex clinical phenotype consisting of ID, severe behavioral problems, and movement abnormalities, including ataxia and spasticity among others, and were found to carry a known pathogenic mutation (p.Val667Met) in the POLR3B (RNA polymerase III subunit B) gene. POLR3B genetic variations are responsible for hypomyelinating leukodystrophy-8 that manifested with similar clinical symptoms as those observed in our family11. The only affected member from family Fam-04 was found to carry a known pathogenic mutation (p.Arg1922/1961Stop) in the vacuolar protein sorting 13 homolog A (VPS13A) gene, mutations of which are responsible for choreoacanthocytosis (CHAC)12. During the disease segregation analyses, an additional sibling (II-2) in this family, who remains unaffected at 30 years old, was found to carry the identified homozygous VPS13A mutation. The clinical features of our patient resemble those described in other reported CHAC patients. Both available patients from family Fam-05 were found to share 11 small tracks of homozygosity (~1Kb-1.5Kb), in which no homozygous genetic variations, including single nucleotide variations (SNVs) or indels, were identified. No compound heterozygous mutations were identified in both affected subjects. By contrast, a novel hemizygous mutation was identified in the SYN1 gene (Xp11.3-p11.23), which encodes for synapsin-1 and has been associated with ASD and epileptic syndromes13,14. This novel SYN1 missense genetic variation, which consisted of a c.1259 G > A transition that resulted in p.Arg420Gln amino-acid substitution, was found to be highly conserved among other species and absent in 840 ethnicity-matched control chromosomes and public databases. It was additionally predicted to be pathogenic by several computational programs including Mutation Taster (disease-causing), MutPred (0.568), and CADD (15.73). Our patients with this novel SYN1 mutation presented with ASD-like features and ID without epileptic seizures. The three affected members from Fam-06 were found to carry a known and homozygous pathogenic mutation (p.Met245Valfs*2) in the SPG11 gene, which is the most commonly mutated gene in complex autosomal recessive hereditary spastic paraplegia (AR-HSP), which presents with spastic paraplegia along with other clinical manifestations including ID. Our patients presented with clinical features similar to those found in AR-HSP including the presence of thin corpus callosum15,16.
Lastly, only one family (Family 7) was identified with pathogenic mutations in an unknown disease gene. In this family only patient 1 was subjected to WGS analyses. The patient’s WGS data identified 12 different homozygous missense single-nucleotide variations (SNVs), with two of them located within previously associated homozygous segments. No compound heterozygous variation was identified. These two SNVs were located within NSL1 (p.Arg74Pro) and RTL1 (p.Arg235His) genes. Both RTL1 (retrotransposon-like protein 1) and NSL1 (Kinetochore-associated protein NSL1 homolog that is a component of MIS12 kinetochore complex) mutations were validated in the patient 2, who was a homozygous carrier for both mutations, and both were found to segregate with disease status. The RTL1 genetic variation was found in heterozygosis in both healthy parents and a healthy sibling and found to be not present in an additional healthy sibling. The healthy family members were found to be either heterozygous carriers (n = 3) or non-carriers (n = 1) for the NSL1 genetic variation (Fig. 1). The RTL1 p.Arg275His mutation was not present in either the Iranome or GME variome databases but it is described in the gnomAD database with a frequency of 2.7e-5 (rs926337313) and it was found in homozygosis in two out of the 91 ethnicity-matched control subjects screened by us through Sanger sequencing. The NSL1 p.Arg74Pro mutation is novel and found to be not present in any of the public SNP databases or in any of ethnicity-matched control subjects investigated through Sanger sequencing (n = 91). In addition, the NSL1 p.Arg74Pro mutation with two mutated nucleotides (Fig. 1), affecting a highly conserved amino-acid (Fig. 3A), was predicted pathogenic by various computational methods [Mutation Taster (disease-causing), MutPred (0.624), SIFT (damage), and CADD (14.58)], while the RTL1 mutation was predicted non-pathogenic by most of them [Mutation Taster (polymorphism), MutPred (0.488), and CADD (9.858)]. Both RTL1 and NSL1 genes are expressed in brain tissues, with RTL1 showing lower brain expression, being localized to the substantia nigra and hypothalamus, while NSL1 was found to be more ubiquitously expressed at higher levels in brain and other tissues, with its expression being higher in the spinal cord (https://gtexportal.org/home/). Given the role of kinetochores in neuronal development17, we also searched for protein-protein interactions in the STRING database to explore whether any of the kinetochore-related proteins associated with ID and microcephaly syndromes interacts with NSL1 and found that NSL1 interact with KNL1 (Fig. 3B).

(A) Conservation of the amino-acid arginine at position 74 of the NSL1 protein across other orthologous. (B) Known and predicted protein-interactions of NSL1 protein according to STRING database. Kinetochore proteins that act as components of the essential kinetochore- associated NDC80 complex, which is required for chromosome segregation and spindle checkpoint activity: NUF2, SPC24, and SPC25. Kinetochore proteins that are part of the MIS12 complex, which is required for normal chromosome alignment and segregation and for kinetochore formation during mitosis: MIS12, PMF1-BGLAP (Polyamine-modulated factor 1), DSN1 (Kinetochore-associated protein DSN1 homolog), NSL1 (Kinetochore-associated protein NSL1 homolog). Kinetochore proteins that are essential for spindle-assembly checkpoint signaling and for correct chromosome alignment: CASC5 (KNL1; Kinetochore scaffold 1) and BUB1 (Mitotic checkpoint serine/threonine-protein kinase BUB1).
Functional results
Reduced neurite outgrowth was observed in mutant SYN1 cells
One of the families reported here was shown to have an X-linked disease due to a novel SYN1 mutation. To further assess the deleterious effects of the disease-segregating SYN1 mutation, primary hippocampal neurons were transfected and allowed to express either wild-type (wt) or mutant (m) SYN1 protein (Fig. 4A). The transfected cells were identified by the expression of GFP. A similar transfection efficiency (~30%) was observed between wt and m proteins (Fig. 4B). However, the mutant primary hippocampal neurons showed lower expression levels than wt neurons (P = 0.0004), indicating that the mutant SYN1 protein led to a significantly reduced neurite outgrowth (Fig. 4C,D).

Effects of SYN1 R420G mutation on hippocampal neurons. (A) Western blot in HEK293T cells untransfected or transfected with either wild-type of R420G SYN1-V5 mutant confirm expression of both constructs. Original western blot images are provided as Supplementary Material (B) Transfection efficiency on hippocampal neurons was calculated in three independent experiments by counting GFP+ cells. Black solid column shows wild-type SYN1-V5 transfected neurons and solid grey column shows R420G SYN1-V5 mutant transfected neurons C: Microscopy images of hippocampal neurons transfected with either wild-type (upper panel) or mutant (lower panel) SYN1-V5 (Scale bar: 25μM). (D) Neurons transfected with wild-type SYN1-V5 express higher levels of SYN1 (p = 0.0004) and (E) have longer axons (p = 0.0008) than their mutant counterparts. The length of axons and normalized fluorescence were measured using the ImageJ software. The graphs shows the mean normalize fluoresce (upper graph) and the axon length in μM (lower graph) of three independent transfections. Black columns show wild-type SYN1-V5 transfected neurons and solid grey columns show R420G SYN1-V5 mutant transfected neurons. Values represent the means ± SEM. ***p < 0.001; ns = non-significant.
Neurite development is impaired in mutant syn1 hippocampal neurons
Synapsins are well known for their participation in the developing nervous system. Particularly, both SYN1 and SYN2 expressions are increased during the formation of neurites, specification of axons, and synapse formation in mammals18,19, and both are involved in neuronal development, axonogenesis and synaptogenesis20,21, Thus, we examined and analyzed the neurites of syn1 wt and m cells. We observed that the length of the neurites in the mutant syn1 cells was shorter than that observed in their wild-type counterparts and that this difference was statistically significant (Fig. 4C,E).
Discussion
In this study, we reported the clinical features and genetic findings of seven different families suffering from complex NDD syndromes. Taking into consideration the complexity of the genetic architecture and the phenotypic heterogeneity of the NDDs22, we used a combination of HM and WGS approaches to establish the disease-causing genetic variations in the affected pedigrees. My group and others have repeatedly demonstrated how powerful the combination of these two techniques is in identifying disease-associated genetic variations and genes23,24,25,26,27,28,29,30. All families were found to carry pathogenic genetic variations in different disease genes, with six families having genetic variations (4 known and 2 novel) in known genes, while one family carried a pathogenic genetic variation in a novel gene (Table 1). The two novel genetic variations identified in known disease genes, SLC5A2 and SYN1, are likely to be pathogenic, as they were the only disease-segregating mutations identified within the disease-associated loci, and both were absent in ethnicity-matched neurologically normal individuals and public SNP databases. Further, our examined patients presented with similar phenotypic manifestations to those previously reported, including the presence of renal glycosuria in patients with SLC5A2 mutations. Developmental delay and movement abnormalities have also been reported in patients with renal glycosuria and SLC5A2 mutations10. Although seizures were not observed in our patients with SYN1 mutations, SYN1 mutations have been found in individuals with epilepsy, ASD, or both. The mutated amino acid in SYN1 (420aa) is located very close to the Synapsin_C domain (214–416 aa), which is an ATP binding domain that is highly conserved across all the synapsins and is a feature of all splice variants. We, therefore, examined the functional effects of the mutant SYN1 protein by mutating the affected amino acid in hippocampal neurons: we observed significantly reduced neurite outgrowth and impaired axonal development in mutant cells when compared to wild-type counterparts (Fig. 4), thus, supporting its pathogenic effect on the protein function and confirming the role of SYN1 function in neuronal development and axonal outgrowth.
Among the pedigrees examined here, we identified one with a pathogenic genetic variation in a novel gene not previously associated with ID syndromes. In this family, which presented with ID, spastic paraplegia and visual impairments, two genetic variations within two different genes (NSL1 and RTL1) were identified. The genetic variation within the RTL1 gene was found in both public databases and ethnicity-matched control sequenced by us. Furthermore, the RTL1 gene encodes for a retrotransposon-like protein 1, whereas NSL1 encodes for a kinetochore-associated protein that contains two coiled-coil domains that localize to kinetochores, which are chromosome-associated structures that attach to microtubules and mediate chromosome movements during cell division. The kinetochores are broadly conserved from yeasts to humans and their constitutive proteins have an essential post-mitotic function in neuronal development17. Interestingly, mutations in microtubule-related genes, including DYNC1H1 (dynein cytoplasmic 1 heavy chain 1), KIF5C (kinesin family member 5 C), KIF2A (kinesin family member 2 A), and TUBG1 (tubulin gamma 1) have been shown to cause malformations of cortical development associated with severe ID and/or microcephaly31; de-novo truncating mutations in CHAMP1 (chromosome alignment maintaining phosphoprotein 1), a protein that is involved in kinetochore-microtubule attachment, have been identified in patients with syndromic ID and dysmorphic facial features32,33,34; and mutations in KNL1 (CASC5), which is a kinetochore scaffold 1 protein that is also required for creation of kinetochore-microtubule attachments and chromosome segregation, and interacts with NSL1 (Fig. 3), are a cause of autosomal recessive primary microcephaly-4 (MCPH4)35.
Taken together, the following evidence strongly supports the novel NSL1 p.Arg74Pro mutation as the genetic variation responsible for causing disease in our reported family: (1) The role of kinetochore-associated proteins in neuronal development and their associations with ID and microcephaly syndromes, (2) the interaction of NSL1 with other kinetochore proteins responsible for causing ID (Fig. 3B), and (3) the fact that the novel NSL1 p.Arg74Pro mutation is predicted highly pathogenic and is highly expressed in brain tissues. Therefore, we believe that impairments within the NSL1 kinetochore-protein might be associated with the development of complex ID and might affect the interaction with other ID-associated kinetochore proteins leading to the development of NDD.
Despite the fact that all the families presented with ID, the clinical spectrum was different in the majority of them (Table 1). All (n = 5) but two families presented with motor abnormalities along with the ID features. The high frequency of motor abnormalities in our families is not surprising since these and seizures are frequently observed in patients with ID4. Novel genes causing ID and parkinsonism have been also recently identified through both whole exome and genome analyses24,36,37,38, seizures have been reported in patients with parkinsonism39,40, and a high frequency of parkinsonism has been observed among adults with ASD41. Epileptic seizures were also present in families with NAGLU, SLC5A2, POLR3B, and VPS13A mutations (Table 1), and behavioral and psychiatric symptoms were observed in a family with POLR3B mutations (Fam 03). Epilepsy is relatively prevalent in people with ID, being more common in people with ID than in the general population, and its prevalence increases with the severity of disability42. Existing evidence suggest that epileptic seizures and side effects of antiepileptic drugs can directly damage individual’s cognitive ability and physical health through repeated head injury and seizures episodes. In fact, behavior abnormalities and psychiatric disorders occur significantly more often in people with epilepsy and people with ID than in “healthy” subjects42,43. Thus, it is not surprising that four out of the seven families reported here also feature epileptic seizures. To summarize, most of our patients with mutations in known genes presented with similar phenotypic manifestations to those previously reported (Table 1), with the exception of our patients with the NAGLU p.D312N mutation, who did not show any behavioral symptom.
In conclusion, we report the genetic defects, including the identification of a novel kinetochore gene as responsible for ID, and the phenotypic manifestations of seven different families with complex ID syndromes, most of which presented with other comorbidities, including epilepsy, motor abnormalities, psychiatric symptoms, and ASD. Given the fact that the neurodevelopmental disorders can be attributed to mutations at over 1000 loci, it is not surprising that different genes were identified to be mutated in each individual family4. Both the phenotypic and genetic heterogeneities associated with NDD disorders make their differential diagnosis increasingly complex44, and thus, we believe that the combination of HM and WGS is a suitable and fruitful strategy to identify the gene defects in recessive pedigrees with parental consanguinity, as it allows to target genomic regions for the analysis by WGS of a single-family member and to determine if distinct causes are responsible for the co-occurrences of rare phenotypes in the same pedigree.
Materials and Methods
Subjects
Seven different families from the Middle East and featuring complex NDD syndromes were clinically diagnosed and subjected to both HM and WGS analyses. The Wechsler IQ test was used to classify the severity of ID. Six out of seven families were consanguineous with parents being first cousins. 420 DNA samples belonging to ethnicity-matched control subjects from different ethnic populations living in Iran (Arabs, Azeris, Balochs, Kurds, Lurs, Persians) were also available for study. DNA samples from all members were isolated from whole blood using standard procedures. The local ethics committee at the Semnan University of Medical Sciences approved this study, and informed consent according to the Declaration of Helsinki was obtained from all participants.
Genetic approaches
Due to the phenotypic heterogeneity observed in our recruited pedigrees (Table 1) and the parental consanguinity observed in six of them (Fig. 1), both HM and WGS analyses were performed to facilitate the identification of the disease-associated mutations. HM analyses were performed in all the families consisting of more than one affected member (n = 6), while the disease-associated mutation for family Fam-04 that consisted of one affected patient was exclusively determined through WGS analysis.
Homozygosity mapping (HM)
High throughput SNP genotyping was carried out in all available family members of six different families (n = 34) (Fig. 1A) using the HumanOmniExpress Exome arrays v1.3 and the HiScanSQ system (Illumina Inc., San Diego, CA, USA). The GenomeStudio program (GS; Illumina) was used to undertake quality assessments and generate PLINK input reports for HM45. Homozygous segments across all family members in each pedigree were determined as previously described, and only those shared by affected family members but not by healthy subjects were considered as disease-associated loci24,27,29. HM analyses were not carried out in Family ID-Fam04, in which direct WGS to identify the gene defects was performed in the only affected family member.
Whole genome sequencing (WGS)
WGS was carried out at the New York Genome Center (NYGC) in at least two affected family members from each family with the exception of family 04 and family 07 in which only one affected subject was subject to WGS analyses (n = 12). Briefly, sequencing libraries were constructed with the TruSeq PCR-free library kit (Illumina) following the manufacturer’s recommended protocol and sequenced on the Illumina HiSeq X instruments, with 2 × 150 bp paired reads, to a minimum coverage of > 30 × . Sequencing data was processed with NYGC’s automated analysis pipeline, which includes alignment to GRCh37 using BWA-MEM (v0.7.8)46, and further processing with GATK best practices, including the marking of duplicates with Picard (v1.83, http://picard.sourceforge.net) and GATK (v3.2.2)47. Single nucleotide variations (SNVs) and indels were called by using the GATK HaplotypeCaller and were jointly genotyped. Deletions were called by using GenomeSTRiP (v2.0)48 and were jointly called by using 17 HapMap individuals (CEPH Platinum Genomes pedigree). All deletions annotated as PASS in the GenomeSTRiP results were further filtered by using custom scripts to remove redundant calls and breakpoints overlapping repeat regions, or with extensive mapping ambiguity. Annotations of variants included predictions of the effect of nucleotide change on protein sequence using SnpEFF; variant frequencies in different populations from 1000 Genomes project, NHLBI GO Exome Sequencing Project; cross-species conservation scores from PhyloP, Genomic Evolutionary Rate Profiling (GERP), PhastCons; functional prediction scores from Polyphen2, SIFT, and CADD49; and variant disease associations from OMIM, Clinvar, Genetic Association Database (GAD); regulatory annotations from ENCODE, RegulomeDB, ORegAnno, KEGG pathway annotations; transcription factor binding sites from the Transfac database; and Gene Ontology (GO) annotations for biological process, cellular component, molecular function.
Selection of candidates: Identification of disease-causing variations
Because in recessive pedigrees, healthy parents are obligate heterozygous carriers for the mutant allele, only homozygous and compound-heterozygous genetic variations were considered as potential disease-causing mutations. The combination of HM and WGS analyses allowed us to further reduce the number of candidate genetic variations in each individual pedigree. Only the genetic variations within the disease-associated chromosomal regions were potentially considered as disease-associated mutations. Thus, only novel and rare (MAF <0.0001, <0.01%) non-synonymous missense, nonsense, frame-shift, gain or loss of stop codon genetic variations within the disease-associated loci were examined further (Fig. 2)24,50,51. To assist with gene identification, the frequency of selected candidates was examined in additional public databases, including the Iranome database, which contains whole exome data of 800 individuals from eight major ethnic groups in Iran (http://www.iranome.com/about), the Greater Middle-East variome that contains exome data from 2,497 individuals (http://igm.ucsd.edu/gme/), and the Genome Aggregation database that contains data from 125,748 exome sequences and 15,708 whole-genome sequences (gnomAD; https://gnomad.broadinstitute.org/). The pathogenicity of the novel identified mutations was predicted by two additional computational methods (MutPred and SNPs&GO) that have been evaluated as most efficient52 and were not included in the annotation files (Fig. 2).
Variant validation and disease segregation assays
Sanger sequencing using primers flanking the exons where the disease-associated mutations were located (NAGLU exon 5, SLC5A2 exon 9, POLR3B exon 19, VPS13A exon 45, SYN1 exon 10, SPG11 exon 4, NSL1 exon 1, and RTL1 exon 1) was used to validate the identified mutations and to perform the disease segregation analyses. Primer sequences were designed by using a public primer design website (http://ihg.gsf.de/ihg/ExonPrimer.html) (Primers sequences available upon request). All purified PCR products were sequenced in both forward and reverse directions with Applied Biosystems BigDye Terminator v3.1 sequencing chemistry as per the manufacturer’s instructions, and resolved and analyzed as described elsewhere27.
Functional assays
SYN1 in-vitro assays
The effects of the wild-type (wt) and mutant (m) amino-acids of a novel SYN1 mutation (p.Arg420Gln) identified in a patient with ID and autistic-like features were examined through in-vitro assays. The SYN1 gene encodes two different isoforms: the isoform SYN1a (NP_598006.1) with 705 amino acids and the isoform SYN1b (NP_008881.2) containing 669 amino acids. Both isoforms are exclusively expressed in brain tissues, with transcript SYN1a expressed at higher levels.
For site-directed mutagenesis and cloning, The pANT7_cGST (GST-tagged in vitro expression vector) SYN1b (Homo sapiens) plasmid was obtained from DNASU Plasmid Repository (Clone ID HsCD00639921) and deposited from the Center for Personalized Diagnostics at Arizona State University (http://dnasu.org/)53. The plasmid was expanded using a Qiagen Spin MiniPrep Kit (Qiagen, Germany). Once validated, a mutation affecting the 420 amino-acid (p.R420G) was introduced by using the QuickChange II XL Site Directed Mutagenesis Kit (Agilent Technologies, USA) following the manufacturer’s recommendations. Primers used to generate the mutation were designed by using QuickChange Primer Design Tool from Agilent Technologies, and primers used to verify the mutation were designed using Primer 3 program (http://bioinfo.ut.ee/primer3-0.4.0/). All constructs were verified in both forward and reverse directions through Sanger sequencing. Subsequently, SYN1 wt and m cDNA in pANT7_cGST-SYN1 plasmid were amplified using corresponding primers (primer sequences available upon request). To generate the longer SYN1a transcript, two different pairs of primers were designed and used to amplify two PCR products containing respectively the 669 amino-acids of SYN1b and the 36 missing amino-acids of SYN1a. Both PCR fragments were then joined and amplified by overlapping PCR, sub-cloned into pcDNA3.1V5/HisA vector (Clontech, Mountain View, CA, USA) between EcoR1 and Xba1 sites, and verified by Sanger sequencing. Their expressions were confirmed by transfecting them into HEK293T cells and immunoblotting the lysates with anti-V5 antibody (ThermoFisher, R960-25).
For cell transfections, HEK293T were maintained in 1X Dubelcco’s Modified Eagle’s Medium (1x DMEM) (Corning, USA), supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin/Streptomycin (GIBCO, USA), and kept in a 37 °C/5%CO2 incubator. Primary hippocampal neuron cultures were prepared from E15.5 wt C57BL6 mouse embryo as previously described54, maintained in neurobasal medium supplemented with B27 and 1% Penicillin/Streptomycin, and kept in a 37 °C/5%CO2 incubator. Both HEK293T cells and hippocampal neurons were plated in 24-well plates with a density around 2*10E05 cells/ml and transfected by using Lipofectamine 3000 (ThemoFisher, USA) according to the manufacturer’s instructions. Briefly, 1 µg of plasmid and 3 µl of Lipofectamine separately diluted in 100 µl of Opti-MEM (GIBCO, USA) were mixed and incubated for 15 min. The 200 µl of plasmid:lipofectamine complex was then added to cells drop by drop and mixed by gently agitation. Transfection efficiency was measured by the number of cells expressing green fluorescent protein (GFP) compared to the total number of cells.
Immunocytochemistry
Non-transfected and transfected hippocampal neurons with either wt or m plasmid were plated on poly-D-lysine coated coverslips at a density of 100/mm2 in a 6-well plate, cultured for 14 days, and fixed by 4% paraformaldehyde treatment. For immunostaining cells were permeabilized with 0.05% Triton X-100 (Sigma Aldrich, USA) for 10 min, blocked with 1% BSA for 30 min, and incubated for 1 hour at room temperature (RT) with primary antibody Anti-V5 tag diluted 1:500 in 1% BSA (Thermo Fisher, USA, R960-25). After washing, fluorophore-conjugated secondary antibody was added for 1 hour at RT (anti-mouse 1:1000 in 1% BSA) (Jackson Immunoresearch, USA, 115-035-166). Cell nuclei were labeled using Hoechst 33342, Trihydrochloride, Trihydrate at 2μg/ml (Thermo Fisher, USA, H3570). Coverslips were mounted into the slides using Fluoromount-G™ Slide Mounting Medium (Electron Microscopy Sciences, USA).
Imaging
Immunolabeled hippocampal neurons were visualized by using a Leica SP5 DM microscope with a 63x magnification objective and analyzed through the ImageJ software (imagej.nih.gov). Confocal z-stack images were acquired in 17 (wt) and 14 (m) random locations within the coverslips of three independent transfections. For SYN1 quantification, individual cells (42 wt and 44 m cells) were circled based on their fluorescent signals and the area and integrated mean intensity was then calculated in ImageJ. The corrected total cell fluorescence (CTCF) was calculated as Integrated Density (area of selected cells x mean fluorescence of background readings)55. The length of the neurites was measured by using the ImageJ software as described elsewhere13.
Western blot
HEK293T cell lysates were collected using the radioactive immunoprecipitation assay (RIPA) buffer (Sigma) with a phosphatase inhibitor (Roche). HEK293T cell lysates were loaded onto NuPAGE 4–12% Bis-Tris Protein Gel (Thermo Fisher, NP0336BOX) and resolved in 1x NuPAGE MES SDS running buffer (Thermo Fisher, NP0002). Proteins were then transferred to PVDF membrane (Thermo Fisher, LC2002) using 1x transfer buffer containing 20% methanol. The membrane was blocked with 10% non-fat dry milk (LabScientific, 732-291-1940) in 1x Tris buffered saline containing 0.05% Tween (TBST) for 30 min and incubated with primary antibody against V5 (1:1000, Thermo Fisher, R960-25) overnight. The membrane was washed three times with 1x TBS, once with 10% non-fat dry milk, and incubated with horseradish peroxidase conjugated anti-mouse secondary antibody (1:5000, Jackson Immunoresearch, 115-035-166) for 30 min. The membrane was washed three times with 1 s TBST and chemiluminescent signal was developed by incubating the membrane with HRP substrate (SuperSignal West Dura, Thermo Fisher, 34075) and detected by G:BOX Chemi image analyzer (Syngene, USA). The membrane was stripped of anti-V5 antibody by incubating with 50 mM Glycine buffer, pH 2.2 for 30 min, and immunoblot procedure was repeated with anti-actin (1:1000, Sigma, A2066) and HRP conjugated anti-rabbit (1:5000, Jackson ImmunoResearch, 711-035-152) antibodies as described above.
Statistical analyses
Statistical analyses were performed using the Mann-Whitney nonparametric test in the GraphPad Prism software version 6.0 (GraphPad, USA). Data on graphs are presented as mean ± SEM. ***P < 0001; ns = non-significant.
Data availability
The raw data that support the findings of this study are now available from the corresponding author upon reasonable request, and will be deposited in dbGAP upon publication.
References
Daily, D. K., Ardinger, H. H. & Holmes, G. E. Identification and evaluation of mental retardation. Am Fam Physician 61(1059–1067), 1070 (2000).
De Rubeis, S. & Buxbaum, J. D. Genetics and genomics of autism spectrum disorder: embracing complexity. Hum Mol Genet 24, R24–31, https://doi.org/10.1093/hmg/ddv273 (2015).
Rapin, I. A. N Engl J Med 337, 97–104, https://doi.org/10.1056/NEJM199707103370206 (1997).
Tarlungeanu, D. C. & Novarino, G. Genomics in neurodevelopmental disorders: an avenue to personalized medicine. Exp Mol Med 50, 100, https://doi.org/10.1038/s12276-018-0129-7 (2018).
Firth, H. V., Wright, C. F. & Study, D. D. D. The Deciphering Developmental Disorders (DDD) study. Dev Med Child Neurol 53, 702–703, https://doi.org/10.1111/j.1469-8749.2011.04032.x (2011).
Buxbaum, J. D. et al. The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron 76, 1052–1056, https://doi.org/10.1016/j.neuron.2012.12.008 (2012).
RK, C. Y. et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20, 602–611, https://doi.org/10.1038/nn.4524 (2017).
Heron, B. et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A 155A, 58–68, https://doi.org/10.1002/ajmg.a.33779 (2011).
Froukh, T. J. Next Generation Sequencing and Genome-Wide Genotyping Identify the Genetic Causes of Intellectual Disability in Ten Consanguineous Families from Jordan. Tohoku J Exp Med 243, 297–309, https://doi.org/10.1620/tjem.243.297 (2017).
van den Heuvel, L. P., Assink, K., Willemsen, M. & Monnens, L. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet 111, 544–547, https://doi.org/10.1007/s00439-002-0820-5 (2002).
Wolf, N. I. et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 83, 1898–1905, https://doi.org/10.1212/WNL.0000000000001002 (2014).
Ogawa, I. et al. Ophthalmologic involvement in Japanese siblings with chorea-acanthocytosis caused by a novel chorein mutation. Parkinsonism Relat Disord 19, 913–915, https://doi.org/10.1016/j.parkreldis.2013.05.012 (2013).
Fassio, A. et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet 20, 2297–2307, https://doi.org/10.1093/hmg/ddr122 (2011).
Garcia, C. C. et al. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet 41, 183–186 (2004).
Paisan-Ruiz, C., Nath, P., Wood, N. W., Singleton, A. & Houlden, H. Clinical heterogeneity and genotype-phenotype correlations in hereditary spastic paraplegia because of Spatacsin mutations (SPG11). Eur J Neurol 15, 1065–1070, https://doi.org/10.1111/j.1468-1331.2008.02247.x (2008).
Paisan-Ruiz, C., Dogu, O., Yilmaz, A., Houlden, H. & Singleton, A. SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70, 1384–1389, https://doi.org/10.1212/01.wnl.0000294327.66106.3d (2008).
Zhao, G., Oztan, A., Ye, Y. & Schwarz, T. L. Kinetochore Proteins Have a Post-Mitotic Function in Neurodevelopment. Dev Cell 48, 873–882 e874, https://doi.org/10.1016/j.devcel.2019.02.003 (2019).
Cesca, F., Baldelli, P., Valtorta, F. & Benfenati, F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol 91, 313–348, https://doi.org/10.1016/j.pneurobio.2010.04.006 (2010).
Fornasiero, E. F., Bonanomi, D., Benfenati, F. & Valtorta, F. The role of synapsins in neuronal development. Cell Mol Life Sci 67, 1383–1396, https://doi.org/10.1007/s00018-009-0227-8 (2010).
Chin, L. S., Li, L., Ferreira, A., Kosik, K. S. & Greengard, P. Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci USA 92, 9230–9234 (1995).
Corradi, A. et al. SYN2 is an autism predisposing gene: loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum Mol Genet 23, 90–103, https://doi.org/10.1093/hmg/ddt401 (2014).
Klein, M., van Donkelaar, M., Verhoef, E. & Franke, B. Imaging genetics in neurodevelopmental psychopathology. Am J Med Genet B Neuropsychiatr Genet 174, 485–537, https://doi.org/10.1002/ajmg.b.32542 (2017).
Paisan-Ruiz, C. et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord 25, 1791–1800, https://doi.org/10.1002/mds.23221 (2010).
Khodadadi, H. et al. PTRHD1 (C2orf79) mutations lead to autosomal-recessive intellectual disability and parkinsonism. Mov Disord 32, 287–291, https://doi.org/10.1002/mds.26824 (2017).
Paisan-Ruiz, C. et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 65, 19–23, https://doi.org/10.1002/ana.21415 (2009).
Taghavi, S. et al. A Clinical and Molecular Genetic Study of 50 Families with Autosomal Recessive Parkinsonism Revealed Known and Novel Gene Mutations. Mol Neurobiol, https://doi.org/10.1007/s12035-017-0535-1 (2017).
Krebs, C. E. et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat 34, 1200–1207, https://doi.org/10.1002/humu.22372 (2013).
Pourreza, M. R. et al. Applying Two Different Bioinformatic Approaches to Discover Novel Genes Associated with Hereditary Hearing Loss via Whole-Exome Sequencing: ENDEAVOUR and HomozygosityMapper. Adv Biomed Res 7, 141, https://doi.org/10.4103/abr.abr_80_18 (2018).
Vahidnezhad, H., Youssefian, L., Jazayeri, A. & Uitto, J. Research Techniques Made Simple: Genome-Wide Homozygosity/Autozygosity Mapping Is a Powerful Tool for Identifying Candidate Genes in Autosomal Recessive Genetic Diseases. J Invest Dermatol 138, 1893–1900, https://doi.org/10.1016/j.jid.2018.06.170 (2018).
Wang, N. H. et al. Homozygosity Mapping and Whole-Genome Sequencing Links a Missense Mutation in POMGNT1 to Autosomal Recessive Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 57, 3601–3609, https://doi.org/10.1167/iovs.16-19463 (2016).
Poirier, K. et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 45, 639–647, https://doi.org/10.1038/ng.2613 (2013).
Tanaka, A. J. et al. De novo pathogenic variants in CHAMP1 are associated with global developmental delay, intellectual disability, and dysmorphic facial features. Cold Spring Harb Mol Case Stud 2, a000661, https://doi.org/10.1101/mcs.a000661 (2016).
Isidor, B. et al. De Novo Truncating Mutations in the Kinetochore-Microtubules Attachment Gene CHAMP1 Cause Syndromic Intellectual Disability. Hum Mutat 37, 354–358, https://doi.org/10.1002/humu.22952 (2016).
Hempel, M. et al. De Novo Mutations in CHAMP1 Cause Intellectual Disability with Severe Speech Impairment. Am J Hum Genet 97, 493–500, https://doi.org/10.1016/j.ajhg.2015.08.003 (2015).
Genin, A. et al. Kinetochore KMN network gene CASC5 mutated in primary microcephaly. Hum Mol Genet 21, 5306–5317, https://doi.org/10.1093/hmg/dds386 (2012).
Wilson, G. R. et al. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with alpha-synuclein pathology. Am J Hum Genet 95, 729–735, https://doi.org/10.1016/j.ajhg.2014.10.015 (2014).
Vauthier, V. et al. Homozygous deletion of an 80 kb region comprising part of DNAJC6 and LEPR genes on chromosome 1P31.3 is associated with early onset obesity, mental retardation and epilepsy. Mol Genet Metab 106, 345–350, https://doi.org/10.1016/j.ymgme.2012.04.026 (2012).
Ramser, J. et al. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum Mol Genet 14, 1019–1027, https://doi.org/10.1093/hmg/ddi094 (2005).
Pandelia, M. E. et al. Substrate-triggered addition of dioxygen to the diferrous cofactor of aldehyde-deformylating oxygenase to form a diferric-peroxide intermediate. J Am Chem Soc 135, 15801–15812, https://doi.org/10.1021/ja405047b (2013).
Koroglu, C., Baysal, L., Cetinkaya, M., Karasoy, H. & Tolun, A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord 19, 320–324, https://doi.org/10.1016/j.parkreldis.2012.11.006 (2013).
Starkstein, S., Gellar, S., Parlier, M., Payne, L. & Piven, J. High rates of parkinsonism in adults with autism. J Neurodev Disord 7, 29, https://doi.org/10.1186/s11689-015-9125-6 (2015).
Bowley, C. & Kerr, M. Epilepsy and intellectual disability. J Intellect Disabil Res 44(Pt 5), 529–543, https://doi.org/10.1046/j.1365-2788.2000.00270.x (2000).
van Ool, J. S. et al. A systematic review of neuropsychiatric comorbidities in patients with both epilepsy and intellectual disability. Epilepsy Behav 60, 130–137, https://doi.org/10.1016/j.yebeh.2016.04.018 (2016).
Kvarnung, M. & Nordgren, A. Intellectual Disability & Rare Disorders: A Diagnostic Challenge. Adv Exp Med Biol 1031, 39–54, https://doi.org/10.1007/978-3-319-67144-4_3 (2017).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575, https://doi.org/10.1086/519795 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498, https://doi.org/10.1038/ng.806 (2011).
Handsaker, R. E., Korn, J. M., Nemesh, J. & McCarroll, S. A. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet 43, 269–276, https://doi.org/10.1038/ng.768 (2011).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315, https://doi.org/10.1038/ng.2892 (2014).
Darvish, H. et al. A novel PUS7 mutation causes intellectual disability with autistic and aggressive behaviors. Neurol Genet 5, e356, https://doi.org/10.1212/NXG.0000000000000356 (2019).
Taghavi, S. et al. A Clinical and Molecular Genetic Study of 50 Families with Autosomal Recessive Parkinsonism Revealed Known and Novel Gene Mutations. Mol Neurobiol 55, 3477–3489, https://doi.org/10.1007/s12035-017-0535-1 (2018).
Thusberg, J., Olatubosun, A. & Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat 32, 358–368, https://doi.org/10.1002/humu.21445 (2011).
Seiler, C. Y. et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res 42, D1253–1260, https://doi.org/10.1093/nar/gkt1060 (2014).
Kajiwara, Y. et al. The human-specific CASP4 gene product contributes to Alzheimer-related synaptic and behavioural deficits. Hum Mol Genet 25, 4315–4327, https://doi.org/10.1093/hmg/ddw265 (2016).
Cappell, K. M. et al. Multiple cancer testis antigens function to support tumor cell mitotic fidelity. Mol Cell Biol 32, 4131–4140, https://doi.org/10.1128/MCB.00686-12 (2012).
Acknowledgements
We thank all the patients and relatives for their cooperation in this study. Microscopy and image analyses were performed at the Microscopy CORE at the Icahn School of Medicine at Mount Sinai. This work was in part supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (R01NS079388; CPR), and the Semnan University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
H.D. and C.P.-R. contributed to the conception and design of the study. H.D. oversaw all clinical assessments and C.P.-R. oversaw all data collection and analysis. L.J.A., A.T., R.M., A.A., E.S., A.H., E.A., A.H.J., B.E., F.J., M.C., J.J. and C.P.-R. contributed to the acquisition, analysis, and interpretation of data. C.P.-R. drafted the manuscript and revised it critically for important intellectual content. All Authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Darvish, H., Azcona, L.J., Tafakhori, A. et al. Phenotypic and genotypic characterization of families with complex intellectual disability identified pathogenic genetic variations in known and novel disease genes. Sci Rep 10, 968 (2020). https://doi.org/10.1038/s41598-020-57929-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57929-4
This article is cited by
-
Trio-based exome sequencing reveals a high rate of the de novo variants in intellectual disability
European Journal of Human Genetics (2022)
-
Familial SYN1 variants related neurodevelopmental disorders in Asian pediatric patients
BMC Medical Genomics (2021)
-
Intellectual disability genomics: current state, pitfalls and future challenges
BMC Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
