
Research Briefing |
Featured
-
-
Article
| Open Access φ-Aromaticity in prismatic {Bi6}-based clusters
φ-Aromaticity in prismatic {Bi6}-based clusters
Aromaticity is a ubiquitous concept in organic chemistry yet it is less widespread for inorganic species. Now the cluster [(CpRu)3Bi6]–, obtained as part of a soluble salt, has been shown to exhibit aromatic behaviour referred to as φ-type, owing to a highly regular {Bi6} substructure causing a non-localizable molecular orbital of \(f_{z^3}\)-like symmetry.
- Benjamin Peerless
- , Andreas Schmidt
- & Stefanie Dehnen
-
Article |
 Glory scattering in deeply inelastic molecular collisions
Glory scattering in deeply inelastic molecular collisions
Molecular energy transfer is thought to follow a simple rule of thumb: high energy transfer requires hard collisions that result in backscattering. However, now it has been observed that an unexpected forward scattering occurs in NO–CO and NO–HD collisions even for high energy transfer. This is attributed to ‘hard-collision glory scattering’, a mechanism that appears to be ubiquitous in molecule–molecule collisions.
- Matthieu Besemer
- , Guoqiang Tang
- & Tijs Karman
-
Q&A |
 Artificial, augmented and automated chemistry
Artificial, augmented and automated chemistry
Jeremy Frey, professor of physical chemistry at the University of Southampton and principal investigator for the AI3SD Network+, talks with Nature Chemistry about the perils of uncertainty in the quality of machine learning data and the synergies between AI and other technologies.
- Russell Johnson
-
Article |
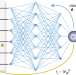 Deep-neural-network solution of the electronic Schrödinger equation
Deep-neural-network solution of the electronic Schrödinger equation
High-accuracy quantum chemistry methods struggle with a combinatorial explosion of Slater determinants in larger molecular systems, but now a method has been developed that learns electronic wavefunctions with deep neural networks and reaches high accuracy with only a few determinants. The method is applicable to realistic chemical processes such as the automerization of cyclobutadiene.
- Jan Hermann
- , Zeno Schätzle
- & Frank Noé
-
Article |
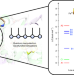 Electronic landscape of the P-cluster of nitrogenase as revealed through many-electron quantum wavefunction simulations
Electronic landscape of the P-cluster of nitrogenase as revealed through many-electron quantum wavefunction simulations
The electronic structures of the metal cofactors of nitrogenase are key to biological nitrogen fixation; however, the [Fe8S7] P-cluster and FeMo cofactor have eluded detailed electronic characterization. Now, the electronic structure of the P-cluster of nitrogenase has been revealed at the many-electron level through exhaustive quantum wavefunction simulations.
- Zhendong Li
- , Sheng Guo
- & Garnet Kin-Lic Chan
-
Article |
 Enhanced reactivity of fluorine with para-hydrogen in cold interstellar clouds by resonance-induced quantum tunnelling
Enhanced reactivity of fluorine with para-hydrogen in cold interstellar clouds by resonance-induced quantum tunnelling
The F + para-H2 → HF + H reaction is an important source of HF in interstellar clouds; however, its unusually high rate and its dynamics at low temperature are not fully understood. Now, quantum-state resolved crossed-beam scattering measurements and anion photoelectron spectroscopy have revealed that this reactivity is caused by a resonance-enhanced tunnelling effect involving a post-barrier resonance state.
- Tiangang Yang
- , Long Huang
- & Daniel M. Neumark
-
Article |
 Relativistic quantum chemical calculations show that the uranium molecule U2 has a quadruple bond
Relativistic quantum chemical calculations show that the uranium molecule U2 has a quadruple bond
Establishing a fundamental understanding of the electronic structure of actinides remains a challenging task for both experiment and theory. Now, it is shown that for the uranium dimer, relativity and electron correlation affects not only the nature of the electronic ground state, but also lowers the bond multiplicity in comparison to previous studies.
- Stefan Knecht
- , Hans Jørgen Aa. Jensen
- & Trond Saue
-
Thesis |
 Talking to Pauling’s ghost
Talking to Pauling’s ghost
Michelle Francl dusts off Pauling’s notes on bonding to explore the illusory link between electron promotion and hybridization.
- Michelle Francl
-
Article |
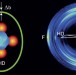 Direct observation of forward-scattering oscillations in the H+HD→H2+D reaction
Direct observation of forward-scattering oscillations in the H+HD→H2+D reaction
Our understanding of reaction dynamics has developed as more accurate measurements of product state-resolved angular distributions have become available. Now, fast forward-scattering oscillations in the product angular distribution of the benchmark chemical reaction H + HD → H2 + D have been observed and are in excellent agreement with quantum-mechanical dynamics calculations.
- Daofu Yuan
- , Shengrui Yu
- & Xueming Yang
-
Article |
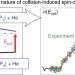 Understanding the quantum nature of low-energy C(3Pj) + He inelastic collisions
Understanding the quantum nature of low-energy C(3Pj) + He inelastic collisions
Collision-induced spin–orbit transitions involve multiple interaction potentials and are by nature non-adiabatic, complicating both their experimental and theoretical study. Crossed-beam experiments and non-Born–Oppenheimer quantum calculations for inelastic collisions of carbon atoms with helium atoms, down to energies corresponding to temperatures below 10 K, have now been performed. Quantum-dynamical resonances predicted by theory were experimentally detected.
- Astrid Bergeat
- , Simon Chefdeville
- & François Lique
-
Article |
 Scattering resonances in bimolecular collisions between NO radicals and H2 challenge the theoretical gold standard
Scattering resonances in bimolecular collisions between NO radicals and H2 challenge the theoretical gold standard
Calculations at the theoretical gold standard generally yield accurate results for a variety of energy-transfer processes in molecular collisions. Using anti-seeding methods in a crossed-beam inelastic scattering experiment, a resonance structure is clearly resolved for NO–H2 collisions, pushing the required accuracy for theoretical potentials beyond the gold standard.
- Sjoerd N. Vogels
- , Tijs Karman
- & Sebastiaan Y. T. van de Meerakker
-
News & Views |
 Caught in the act
Caught in the act
Femtochemistry, the real-time study of reactions on a timescale that captures the molecular and atomic activity involved, has traditionally been performed in the gas or liquid phase. It has now been extended to the solid state in a study that highlights how a controlled reaction environment can place steric constraints on the motions of photoproducts.
- Giulio Cerullo
- & Marco Garavelli
-
Article |
 Coherent ultrafast lattice-directed reaction dynamics of triiodide anion photodissociation
Coherent ultrafast lattice-directed reaction dynamics of triiodide anion photodissociation
Dissociative reactions in the solid state are prone to sample damage. Now, improved sample handling and measurement conditions enable the study of the dissociative reaction of a model triatomic system in the solid state on ultrafast timescales, revealing the significant impact of lattice coordination on the reaction pathway.
- Rui Xian
- , Gastón Corthey
- & R. J. Dwayne Miller
-
Article |
 Wide-dynamic-range kinetic investigations of deep proton tunnelling in proteins
Wide-dynamic-range kinetic investigations of deep proton tunnelling in proteins
A temperature-dependent kinetic study of ground-state proton transfer in the green fluorescent protein highlights the role of ‘deep tunnelling’ in proton wires. A potential mechanism for directional proton transport is proposed, where high-pKa amino acid residues act as ‘tunnel diodes’ and as stabilizing elements within protein water wires.
- Bridget Salna
- , Abdelkrim Benabbas
- & Paul M. Champion
-
Article |
 Molecular hydrogen interacts more strongly when rotationally excited at low temperatures leading to faster reactions
Molecular hydrogen interacts more strongly when rotationally excited at low temperatures leading to faster reactions
The rotational state of a molecule is not generally considered to play a role in how fast it reacts; however, when the temperature is low quantum effects become more important. Now, it is shown that at low temperatures rotationally excited H2 molecules react with He faster than non-rotating ground-state molecules — a process mediated by stronger long-range attraction.
- Yuval Shagam
- , Ayelet Klein
- & Edvardas Narevicius
-
Feature |
 Quantum reform
Quantum reform
Quantum computers potentially offer a faster way to calculate chemical properties, but the exact implications of this speed-up have only become clear over the last year. The first quantum computers are likely to enable calculations that cannot be performed classically, which might reform quantum chemistry — but we should not expect a revolution.
- Leonie Mueck
-
Article |
 Low-energy spectrum of iron–sulfur clusters directly from many-particle quantum mechanics
Low-energy spectrum of iron–sulfur clusters directly from many-particle quantum mechanics
FeS clusters are a universal motif in organisms and are central to many processes, including nitrogen fixation and respiration. By carrying out the first many-electron calculation of the [2Fe-2S] and [4Fe-4S] clusters, they are shown to have an unusual set of closely packed energy levels, which are key to understanding their reactivity.
- Sandeep Sharma
- , Kantharuban Sivalingam
- & Garnet Kin-Lic Chan
-
News & Views |
 Isotope effects feel the cold
Isotope effects feel the cold
Kinetic isotope effects are widely used to elucidate reaction mechanisms and are generally interpreted in terms of simple kinetic models. Measurements of this effect for the Penning ionization reaction between helium and dihydrogen highlight the need for a quantum description of chemical reaction rates when sub-kelvin temperatures are approached.
- Mark Brouard
-
Article |
 Observation of the isotope effect in sub-kelvin reactions
Observation of the isotope effect in sub-kelvin reactions
In cold chemistry, quantum phenomena in reactants' translational motion lead to the temporary trapping of reactants in a collisional complex. It is now shown that this metastable complex is responsible for a dramatic quantum kinetic isotope effect as observed in Penning ionization reactions at low temperatures.
- Etay Lavert-Ofir
- , Yuval Shagam
- & Edvardas Narevicius
-
-
News & Views |
 A multitude of spins
A multitude of spins
Accurately representing molecules with many coupled unpaired electrons is currently impossible using conventional electronic-structure theories. Now, using a recently developed approach, the near-exact quantum wavefunction of the highly complex Mn4CaO5 cluster of photosystem II has been calculated.
- Jeremy N. Harvey
-
Article |
 Entangled quantum electronic wavefunctions of the Mn4CaO5 cluster in photosystem II
Entangled quantum electronic wavefunctions of the Mn4CaO5 cluster in photosystem II
Many-electron quantum modelling of the metal clusters in metalloenzymes is a long-standing ambition for theoreticians. Here, using the density matrix renormalization group, the many-electron wavefunctions of the Mn4CaO5 cluster of photosystem II are computed, providing new insights into the electronic structure and reactivity at the level of many-particle quantum mechanics and entanglement.
- Yuki Kurashige
- , Garnet Kin-Lic Chan
- & Takeshi Yanai
-
Article |
 Ab initio carbon capture in open-site metal–organic frameworks
Ab initio carbon capture in open-site metal–organic frameworks
Metal–organic frameworks featuring unsaturated metal sites have emerged as promising materials for CO2 capture, but the host–guest interactions at play have remained poorly understood. An approach based on quantum chemical calculations has now been devised to generate force fields that accurately describe a MOF's metal sites and predict its gas uptake abilities.
- Allison L. Dzubak
- , Li-Chiang Lin
- & Laura Gagliardi
-
News & Views |
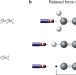 Quadruply bonded carbon
Quadruply bonded carbon
Determining molecular bond orders can be a delicate and sophisticated task, especially if the electronic structure of the studied system is complex. Now, two different ab initio methods have revealed that C2 and analogous species have a fourth bond, rather than the previously assumed maximum of three.
- Jörg Grunenberg
-
Thesis |
 A quantum of history
A quantum of history
Michelle Francl wonders how much time chemists should spend learning history.
- Michelle Francl
-
Article |
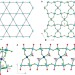 An ionothermally prepared S = 1/2 vanadium oxyfluoride kagome lattice
An ionothermally prepared S = 1/2 vanadium oxyfluoride kagome lattice
Candidates for 'quantum spin liquid' materials are rare and often composed of two-dimensional kagome arrays of d9 centres. Analogous systems based on d1 metal ions may confer different properties, but there are no previously known examples. An inorganic–organic hybrid vanadium d1 material has now been prepared that seems to be an excellent candidate for a spin-liquid ground state.
- Farida H. Aidoudi
- , David W. Aldous
- & Philip Lightfoot
-
News & Views |
 Beyond billiard-ball collisions
Beyond billiard-ball collisions
The collision of an atom and a diatomic molecule may sound like a simple process but it has long been studied to understand the inherent intricacies of collisional energy transfer. Now, experiments carried out in unprecedented detail on the scattering of NO by Ar have revealed further complexity: parity-dependent quantum interference effects.
- David W. Chandler
-
Article |
 Interference structures in the differential cross-sections for inelastic scattering of NO by Ar
Interference structures in the differential cross-sections for inelastic scattering of NO by Ar
Differential cross sections for the rotationally inelastic scattering of NO by Ar are reported with unprecedented quantum-state resolution. The experiments give important details about the mechanism of this fundamental collisional process, providing evidence for a parity-dependent quantum-mechanical interference effect.
- C. J. Eyles
- , M. Brouard
- & S. Stolte
-
Article |
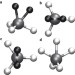 Quantum-induced symmetry breaking explains infrared spectra of CH5+ isotopologues
Quantum-induced symmetry breaking explains infrared spectra of CH5+ isotopologues
Stepwise deuteration of protonated methane CH5+ — a fluxional structure that undergoes ‘hydrogen scrambling’ — leads to dramatic changes in the infrared spectra of the isotopologues. The spectra can be assigned using ab initio quantum simulations that account for the non-classical occupation — by H and D atoms — of topologically different sites within the molecule.
- Sergei D. Ivanov
- , Oskar Asvany
- & Stephan Schlemmer
-
News & Views |
 Chemistry from photons
Chemistry from photons
The use of conventional computers to calculate molecular properties is hindered by the exponential increase in computational cost on increasing the size of the molecules studied. Using quantum computers could be the solution and the initial steps are now being taken.
- Kenneth R. Brown
-
Article |
 Towards quantum chemistry on a quantum computer
Towards quantum chemistry on a quantum computer
Precise calculations of molecular properties from first-principles set great problems for large systems because their conventional computational cost increases exponentially with size. Quantum computing offers an alternative, and here the H2 potential energy curve is calculated using the latest photonic quantum computer technology.
- B. P. Lanyon
- , J. D. Whitfield
- & A. G. White
 Hot spots of radiation damage from extensive water ionization around metal ions
Hot spots of radiation damage from extensive water ionization around metal ions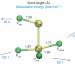 United they are strong
United they are strong