
Abstract
A novel class of geldanamycin (GA) derivatives as hepatitis C virus (HCV) replication inhibitors has been synthesized and their anti-HCV activities were evaluated in GS4.3 HCV replicon cells. Most of the synthesized compounds demonstrated potential activities against HCV in vitro. Substitution with an aliphatic cyclic group (2b) and polar phosphate group (2f) at the 17 position of GA resulted in more potent inhibitory activity. The configurations of the tetrahydrofurfurylamino (THFM) substituents obviously affected their antiviral activities. The 2b with a 2′-(R)-THFM group at the 17 position showed much potent activity and higher selectivity than its 2′-(S) and 2′-(R, S) epimers. In the tested GA derivatives, 2b and 2f show the most potential leading compounds for development of novel anti-HCV agents.
Similar content being viewed by others


Introduction
Approximately 3% of the worldwide population is infected with the hepatitis C virus (HCV),1 but a protective vaccine against HCV does not exist and many patients do not qualify or tolerate standard therapy, which consists of a combination of pegylated interferon (IFN) and ribavirin (RBV).2 Therefore, more effective novel therapeutic strategies are needed.
Recent advances in the understanding of the HCV genome organization, life cycle and the development of HCV replicons and infectious viral particles in tissue culture systems have enabled the rational evaluation of agents that may inhibit HCV replication.3, 4 NS3/4A protease and NS5B polymerase, which are viral enzymes, are currently the most promising targets for discovery of specific HCV inhibitors, and several compounds have already been tested in the clinic, although their ultimate clinical utility is still under investigation.4, 5, 6 Other HCV enzymes, helicase, NS2 protease and NS5A, have also been identified as therapeutic targets.4, 5, 6 However, almost all compounds designed as new therapeutic targets against HCV (STAT-C) have been found to generate drug resistance.4
Recent research has shown that intracellular cofactors are the most promising targets for the design of specific drugs that reduce the resistance to virus.7 Several viruses require viral and host molecular chaperones for entry, replication and assembly, as well as for other steps in viral production.8, 9 Recently, Hsp90 was found to be able to bind to human FKBP8 and form a complex with HCV NS5A. It was identified that the interaction between FKBP8, Hsp90 and HCV NS5A is essential for HCV replication.10 This suggested that Hsp90 may be a promising target to inhibit HCV replication, and inhibition of Hsp90 could provide a feasible therapeutic strategy for the treatment of HCV infection.9, 11 Geldanamycin (GA) and its derivative 17-DMAG, both well known Hsp90 inhibitors, are highly effective in suppressing HCV genome replication in HCV-infected chimeric mice with humanized livers.11 These results indicated that GA should be a good lead compound for developing new agents for combating HCV infectious disease.
In this paper, a series of novel 17-substituted GA derivatives were synthesized and their ability to inhibit HCV replication was evaluated in GS4.3 HCV replicon cells. Their structure–activity relationships against HCV were also investigated.
Results
Chemistry
Different substitutes were connected to positions 17 and/or 19 of the ansa-framework of GA. A total of 13 novel GA derivatives, 11 derivatives substituted with different cyclic groups at the 17 position, one substituted with a phosphate group at the 17 position and one bis-substituted derivative at both the 17 and 19 positions, were synthesized from GA and the corresponding amine.
As shown in Scheme 1, the synthesis of GA derivatives with a single substituted group at the 17 position (compounds 2a–2k) was performed by a one-step reaction of GA with the appropriate amine with or without the presence of a basic reagent such as triethylamine in chloroform.12 To a solution of GA and 2.0 eq Et3N in 20 ml of chloroform, 1.5 eq of amine was added. The reaction mixture was stirred overnight at room temperature and monitored by TLC. When the reaction was completed, the mixture was filtered. The filtrate was concentrated on a rotary evaporator, and the residue was dissolved in ethyl acetate, then they were sequentially washed with water, 1 M aqueous hydrochloric acid, saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride. The organic solution was dried over anhydrous sodium sulfate, filtered and evaporated to dryness, purified by recrystallization with acetone/ether (1/1.2, v/v) and the target compounds were obtained as purple solids (yield 50–80%).
The 17- and 19-bis-substituted derivative 3 was obtained using compound 2 as the starting material following a similar procedure to that used to synthesize the compounds with a single substitution at the 17 position, except that the reaction was maintained for a longer time in the presence of excess amine. All of the products were purified by recrystallization with acetone/ether (1/1.2, v/v).
The preparation of 2l was similar as described in Scheme 2. The substitution at position 17 in compound 2l was composed of nicotinic acid and ethyldiamine, and the synthetic procedures are shown in Scheme 2. To conserve the starting material GA, the amino side group containing the nicotinic acid moiety was synthesized first and this was connected with GA in the last step. Nicotinic acid was reacted with thionyl chloride to obtain the corresponding acyl chloride. The acyl chloride reacted with the N-protected ethyldiamine that produced the N-protected amide. This amide was deprotected to the amino group under acidic conditions. Finally, the nicotinic acid portion was combined with GA to obtain the target product 2l.
The structures and purities of 2a–3 were determined by 1H NMR, MS and HPLC.
Pharmacological results
Effect of GA derivatives on HCV replication in HCV replicon cells
The anti-HCV activity of all synthesized compounds was assessed in GS4. 3 HCV replicon cells, a human hepatoma HuH-7 cell line carrying an HCV subgenomic replicon (Figure 1).11 Intron A (IFN-α-2b) and RBV were used as positive controls, and the results are summarized in Table 1.

Schematic representation of the hepatitis C virus subgenomic replicons used in this study; neo, neomycin phosphotransferase gene and EMCV-I, encephalomyocarditis virus internal ribosomal entry site.
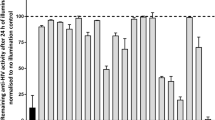
In this work, the IC50 and CC50 of compounds 2a–3 were determined. All of the tested compounds showed inhibitory activity against HCV RNA replication in a dose-dependent manner (Figure 2) in GS4.3 HCV replicon cells. The inhibitory activity of compounds 2b, 2d, 2f, 2g, 2h and 2i were more potent, especially compound 2f (IC50 of 0.0024 μM), which was a better inhibitor of HCV RNA replication than the lead compound GA (IC50 of 0.0048 μM). RBV showed very limited inhibitory activity compared with the lead compound GA in our test. Intron A inhibited HCV RNA replication (IC50 of 4.59 U ml−1) under the test conditions. Bis-substitution at the 17 and 19 positions of GA (compound 3, IC50 is over 58 μM) obviously reduced anti-HCV activity.

The inhibitory activities against hepatitis C virus (HCV) replication under different concentrations of tested compounds in GS4.3 HCV replicon cells. After 72 h of treatment, the number of HCV copies in cells were determined by quantitative real-timePCR and cell viability by the MTT assay. The data represent the mean of three values and the bars indicate the s.d.
The cytotoxicity of most of the tested derivatives was highly decreased compared with the lead compound GA. At the same time, compounds 2b, 2d, 2f, 2h and 2k showed better selectivity than GA. The results also showed that 17-tetrahydrofurfurylamino (THFM) derivatives (2b, 2c and 2d) suppressed replication of the HCV RNA in cells in a configuration-dependent manner (Figure 2). The IC50 against HCV replication in the cells were 0.029 μM for 2b, 0.367 μM for 2c and 0.077 μM for 2d. Compound 2b, with a 2′-(R)-configuration THFM group, showed much stronger inhibitory activity and higher selectivity than its epimer 2c and racemic body 2d. The S-epimer (2c) resulted in more toxicity rather than antiviral activity. The calculated selected indices were 10.8 for 2b, 2.4 for 2c and 6.6 for 2d.
Synergistic inhibition of HCV RNA replication by 2b with Intron A in GS4.3 HCV replicon cells
Because of its critical role at all stages of therapy, pegylated IFN is likely to remain the cornerstone of HCV therapy.2 In any case, drug combinations will be required for treatment of HCV, similar to what has been shown for HIV.6 Therefore, we evaluated the combination of IFN and 2b. Like RBV, 2b exhibited significant synergistic inhibition of HCV RNA replication in the presence of IFN in GS4.3 HCV replicon cells. It is possible that they act synergistically by both decreasing HCV replication and interfering with the ability of HCV to evade the mediators of IFN action.
To ascertain the potential for direct synergy of antiviral effects between Intron A and Hsp90 inhibitor, we selected the relative potent active compound 2b from the synthesized compounds to test its anti-HCV synergistic effects with Intron A in HCV replicon cells. RBV was used as a positive control because it can directly inhibit HCV replication in cells and has synergistic effects with IFN.13, 14 As discussed above similar to RBV, 2b provided a significant improvement in the rate of inhibition of viral replication in comparison with that of Intron A alone (Table 2 and Figure 3).

Anti-hepatitis C virus (HCV) synergistic effects of 2b (a) and ribavirin (b) with Intron A (interferon-α-2b) in GS4.3 HCV replicon cells.
Unlike targeting virus proteins as possible antiviral therapies for HCV (STAT-C), such as protease inhibitors and polymerase inhibitors, 2b targets on the host factor Hsp90, which may not be plagued by the twin problems of viral diversity and escape mutations that interfere with the effectiveness of conventional antiretroviral drugs. Compound 2b may also reduce the required dosage of IFN, which in turn may decrease the adverse effects associated with treatment with IFN. Therefore, 2b may be a promising lead candidate for the development of new antiviral drugs, especially when combined with IFN.
Discussion
SAR analysis
By examination of the relationships between the structure and the anti-HCV activity of the tested compounds, some general conclusions can be drawn as follows: (i) A 17-substituted GA derivative with a phosphate group (2f) showed increased inhibitory activity towards suppressing HCV replication. (ii) Varying the 17 position cyclic substituents exhibits obvious effects on their HCV inhibitory activities. Compounds where the 17 position was substituted with aromatic cyclic groups (that is, 2a, 2e, 2k and 2l) were less potent than where the 17 position was substituted with an aliphatic cyclic group (that is, 2b, 2c, 2d, 2g, 2h, 2i and 2j). (iii) The configuration of the substituent at the 17 position of GA affects their antiviral activities. For 17-THFM GA derivatives, the substituent with a 2′-(R)-THFM group (compound 2b) was a better inhibitor and had higher selectivity than its 2′-(S)-epimer 2c and 2′-(R,S)-diasereomixture 2d. The S-epimer (2c) was found to have increases toxicity rather than increased antiviral activity. (iv) The 17 and 19 position bis-substitutions of GA (3) reduced the anti-HCV activity sharply compared with the 17 position mono-substituted derivatives. (v) This class of compounds possessed synergistic effects with Intron A for inhibiting HCV replication. Compound 2b provided a significant improvement in the rate of inhibiting viral replication in comparison with that of Intron A alone.
In summary, a series of novel GA derivatives were prepared and their inhibitory activities towards HCV replication were evaluated in GS4.3 HCV replicon cells. Most of the synthesized compounds were demonstrated to possess potent activities against HCV in vitro, except 17- and 19-bi-(R)-THFM-substituted compound 3. The selected indices of compounds 2b and 2f were similar to 17-DMAG, one of the effective derivatives on HCV replication.11 The substitution at the 17 position of GA with a polar phosphate group (that is, 2f) and an aliphatic cyclic group (that is, 2b, 2c, 2d, 2g, 2h, 2i and 2j) is helpful for improving anti-HCV activity. The configurations of the substituent at the 17 position can obviously affect their antiviral activities. Consistent with 17-DMAG, this class of compounds possesses synergistic effects with Intron A towards inhibiting HCV replication. Compound 2b provided a significant improvement in the rate of inhibiting viral replication in comparison with that of Intron A alone.
This work demonstrated that several novel anti-HCV agents targeted on Hps90 can be obtained from modifications of GA. Compounds 2b and 2f were similar to 17-DMAG and had especially potential activity towards inhibiting HCV RNA replication compared with the lead compound GA. They both represent potential leads in the development for new anti-HCV drugs.
Methods
General experimental procedures
GA was provided by the Shanghai Institute of Pharmaceutical Industry. Unless noted otherwise, all reagents and solvents were purchased from commercial sources and used without further purification. The 1H NMR spectra were recorded in CDCl3 or CD3OD solution with a Varian Inova 400 or 600 MHz spectrometer (Varian, San Francisco, CA, USA). Chemical shift is reported in parts per million relative to tetramethylsilane as the internal standard. Melting points were determined with a X6 microscope melting point apparatus and were uncorrected. FAB and ESI mass spectra were recorded on an Autospec UltimaTOF mass spectrometer (Micromass UK, Manchester, UK). TLC was performed on precoated aluminum sheets of silica gel 60 F254 (Merck, KGaA, Darmstadt, Germany). Flash chromatography was performed on silica gel, eluted with 1–5% MeOH in dichloromethane. HPLC analysis was performed on a Shimadzu 10Avp HPLC system (Shimadzu Corporation, Tokyo, Japan) equipped with a Shimadzu C18 column (5 μm, 4.6 × 250 mm, Shimadzu Corporation). The mobile phase consisted of water (containing 0.02 mol l−1 KH2PO4) and MeOH (20/80), flowing at a rate of 1.0 ml min−1, and analytes were detected by UV absorption with detection at 332 nm. The percent purity of the tested compounds was >96% using a HPLC area normalization method. Optical rotation activity was measured on a Autopol IV-T Automatic Polarimeter (Rudolph research analytical, Hackettstown, NJ, USA) with detection at 436 nm in anhydrous MeOH at 20 °C, the special optical rotation [α]43620 of GA was +247.7 ° (c=0.0166, CH3OH).
17-[2′-(Thiophen-2′-yl)ethanamine]-17-demethoxygeldanamycin (2a)
To a solution of GA (500 mg, 892.9 μmol) and 2.0 eq Et3N in 20 ml of CHCl3, 1.5 eq of 2′-(thiophen-2′-yl)ethanamine was added. The reaction mixture was stirred overnight at room temperature and monitored by TLC. When the reaction was completed, the mixture was filtered. The filtrate was concentrated on a rotary evaporator, and the residue was dissolved in EtOAc, washed sequentially with water, 1 M aqueous hydrochloric acid, saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride. The organic solution was dried over anhydrous sodium sulfate, filtered and evaporated to dryness, giving a purple solid which was further purified by recrystallization with EtOAc/ether to yield 2a 301 mg (yield 51%) as a purple solid (HPLC purity: 96.7%). m.p. 182–185 °C. [α]20436=+653.6° (c=0.0153, CH3OH). The 1H NMR (400 MHz, CDCl3): δ 0.95–1.00 (m, 6H), 1.72–1.79 (m, 5H), 2.02 (s, 3H), 2.35–2.41 (m, 1H), 2.68–2.75 (m, 2H), 2.87 (s, 2H), 2.95 (s, 2H), 3.16–3.20 (m, 2H), 3.26 (s, 3H), 3.36 (s, 3H), 3.43–3.58 (m, 2H), 3.80 (br, 2H), 4.30 (d, J=10.0 Hz, 1H), 4.84 (br, 2H), 5.18 (s, 1H), 5.83–5.90 (m, 2H), 6.36 (br, 1H), 6.57 (t, J=11.4 Hz, 1H), 6.90–6.98 (m, 2H), 7.21 (d, J=4.8 Hz, 1H), 8.01 (br, 1H) and 9.14 (s, 1H). MS(ESI) m/z calcd. for C34H45N3O8S 655.3; found (M+H+) 656.2.
The following compounds (2b–2k) were obtained in an analogous way.
17-(2′-(R)-THFM)-17-demethoxygeldanamycin (2b)
Compound 2b was synthesized (yield 72.0%) as a purple solid (HPLC purity: 99.3%). [α]43620=+828.4° (c=0.0188, CH3OH). m.p. 135–138°C. The 1H NMR (400 MHz, CDCl3): δ 0.89–1.02 (m, 6H), 1.15 (d, J=10.0 Hz, 2H), 1.38–1.40 (m, 1H), 1.57–1.72 (m, 4H), 1.80 (s, 3H), 1.93–1.99 (m, 2H), 2.03 (s, 3H), 2.34–2.40 (m, 1H), 2.66–2.78 (m, 2H), 3.27 (s, 3H), 3.36 (s, 3H), 3.38–3.47 (m, 1H), 3.54–3.62 (m, 1H), 3.78–3.85 (m, 1H), 3.91–3.95 (m, 1H), 4.08–4.11 (m, 1H), 4.31 (d, J=9.6 Hz, 1H), 4.71 (br, 2H), 5.19 (s, 1H), 5.80–5.92 (m, 2H), 6.54–6.61 (m, 1H), 6.96 (d, J=11.2 Hz, 1H), 7.28 (s, 1H) and 9.17 (br, 1H). MS(FAB) m/z calcd. for C33H47N3O9 629; found (M+Na+) 652.
17-(2′-(S)-THFM)-17-demethoxygeldanamycin (2c)
Compound 2c was synthesized (yield 66%) as a purple solid (HPLC purity: 98.7%). [α]43620=+782.0° (c=0.0160, CH3OH). m.p. 158–161°C. The 1H NMR (400 MHz, CDCl3): δ 0.93–1.16 (m, 6H), 1.25 (s, 2H), 1.32–1.42 (m, 1H), 1.60–1.72 (m, 2H), 1.73–1.78 (m, 2H), 1.80 (s, 3H), 1.91–1.96 (m, 2H), 2.06 (s, 3H), 2.43–2.47 (m, 1H), 2.90–3.02 (m, 2H), 3.23 (s, 3H), 3.39 (s, 3H), 3.27–3.36 (m, 2H), 3.79–3.96 (m, 2H), 4.20–4.37 (m, 2H), 4.97 (s, 1H), 5.45–5.60 (m, 1H), 5.82–6.02 (m, 1H), 6.52–6.62 (m, 1H), 6.95–7.00 (m, 1H) and 7.26–7.31 (m, 1H). MS(FAB) m/z calcd. for C33H47N3O9 629; found (M+Na+) 652.
17-THFM-17-demethoxygeldanamycin (2d)
Compound 2d was synthesized (yield 70%) as a purple solid (HPLC purity: 97.7%). [α]43620=+845.6° (c=0.0177, CH3OH). m.p. 126–129 °C. The 1H NMR (400 MHz, CDCl3): δ 0.90–1.01 (m, 6H), 1.25 (s, 2H), 1.40–1.52 (m, 1H), 1.61–1.65 (m, 2H), 1.70–1.74 (m, 2H), 1.79 (s, 3H), 1.93–1.98 (m, 2H), 2.02 (s, 3H), 2.36–2.39 (m, 1H), 2.66–2.75 (m, 2H), 3.26 (s, 3H), 3.36 (s, 3H), 3.42–3.49 (m, 1H), 3.56–3.62 (m, 1H), 3.79–3.95 (m, 2H), 4.08–4.11 (m, 1H), 4.31 (d, J=10.0 Hz, 1H), 4.80 (br, 1H), 5.19 (s, 1H), 5.85 (t, J=11.2 Hz, 1H), 5.90 (d, J=10.0 Hz, 1H), 6.58 (t, J=11.4 Hz, 1H), 6.95 (d, J=11.6 Hz, 1H), 7.27 (s, 1H) and 9.16 (s, 1H). MS(FAB) m/z calcd. for C33H47N3O9 629; found (M+Na+) 652.
17-(3′,4′-Methylenedioxybenzylamino)-17-demethoxygeldanamycin (2e)
Compound 2e was synthesized (yield 74%) as a purple solid (HPLC purity: 98.4%). [α]43620=+431.2° (c=0.0177, CH3OH). m.p. 206–210°C. The 1H NMR (400 MHz, CDCl3): δ 0.99–1.03 (m, 6H), 1.80 (s, 3H), 2.03 (s, 3H), 2.41–2.47 (m, 1H), 2.68 (d, 1H), 2.73–2.77 (m, 1H), 2.88 (br, 1H), 2.95 (br, 1H), 3.27 (s, 3H), 3.37 (s, 3H), 3.44–3.60 (m, 2H), 4.18 (br, 1H), 4.31 (d, J=10.0 Hz, 1H), 4.48–4.68 (m, 2H), 4.79 (br, 2H), 5.19 (s, 1H), 5.84–5.93 (m, 2H), 5.99 (d, 2H), 6.36 (br, 1H), 6.58 (t, J=11.5 Hz, 1H), 6.73–6.82 (m, 3H), 6.96 (d, J=12.0 Hz, 1H), 7.30 (s, 1H), 8.02 (br, 1H) and 9.16 (s, 1H). MS(ESI) m/z calcd. for C36H45N3O10 679.3; found (M+Na+) 702.2.
17-(1′-Diethylphosphonate-methylamine)-17-demethoxygeldanamycin (2f)
Compound 2f was synthesized (yield 56%) as a purple solid (HPLC purity: 98.4%). [α]43620=+733.3° (c=0.0161, CH3OH). m.p. 174–177 °C. The 1H NMR (300 MHz, CDCl3): δ 0.94–1.04 (m, 6H), 1.36 (m, 6H), 1.60–1.82 (br, 1H), 1.83–1.91 (m, 6H), 1.95–2.02 (m, 3H), 2.18 (s, 1H), 2.31–2.39 (m, 1H), 2.68–2.71 (m, 2H), 3.28 (s, 3H), 3.36 (s, 3H), 3.43 (d, J=9.2 Hz, 1H), 3.58 (d, J=9.2 Hz, 1H), 3.91–4.02 (m, 2H), 4.15–4.21 (m, 4H), 4.33 (d, J=10.0 Hz, 1H), 4.78 (br, 2H), 5.27 (s, 1H), 5.81 (s, 1H), 5.90 (s, 1H), 6.32 (br, 1H), 6.67 (t, J=11.2 Hz, 1H),6.9 (d, J=11.6 Hz, 1H) and 9.07 (s, 1H). MS(FAB) m/z calcd. for C33H50N3O11P 695; found (M+Na+) 718.
17-(2′-(N-ethylpyrrolidinyl)-methylamino)-17-demethoxygeldanamycin (2g)
Compound 2g was synthesized (yield 86%) as a purple solid (HPLC purity: 96.2%). [α]43620=+843.8° (c=0.0159, CH3OH). m.p.106–109 °C. The 1H NMR (400 MHz, CDCl3): δ 0.96–1.01 (m, 5H), 1.09–1.14 (s, 3H), 1.51–1.55 (m, 5H), 1.76–1.80 (m, 2H), 1.81 (s, 3H), 1.90–2.00 (m, 1H), 2.03 (s, 3H), 2.21–2.29 (m, 2H), 2.35–2.46 (m, 1H), 2.65–2.80 (m, 3H), 3.27 (s, 3H), 3.37 (s, 3H), 3.41–3.76 (m, 4H), 4.32 (d, J=10.0 Hz, 1H), 4.51–4.70 (m, 2H), 5.19 (s, 1H), 5.83–5.94 (m, 2H), 6.59 (t, J=11.2 Hz, 1H), 6.96 (d, J=11.2 Hz, 1H), 7.16–7.21 (m, 1H), 7.26–7.32 (m, 1H) and 9.22 (d, J=6.4 Hz, 1H). MS(ESI) m/z calcd. for C35H52N4O8 656.4; found (M+H+) 657.3.
17-(2′-(1′-Oxa-4′-aza-heterocyclohexyl-1′-)ethylamino)-17-demethoxygeldanamycin (2h)
Compound 2h was synthesized (yield 47%) as a purple solid (HPLC purity: 99.8%). [α]43620=+871.2° (c=0.0189, CH3OH). m.p. 125–128 °C. The 1H NMR (400 MHz, CDCl3): δ 0.89–1.03(m, 6H), 1.28–1.38 (m, 2H), 1.42–1.51 (m, 1H), 1.64–1.68 (m, 2H), 1.78 (s, 3H), 2.03 (s, 3H), 2.50–2.61 (m, 4H), 2.64–2.71 (m, 4H), 2.70–2.81 (m, 1H), 3.27 (s, 3H), 3.36 (s, 3H), 3.44 (d, J=9.2 Hz, 1H), 3.54 (br, 1H), 3.58 (d, J=9.2 Hz, 1H), 3.64–3.80 (m, 4H), 4.31 (d, J=10.0 Hz, 1H), 4.41 (br, 1H), 4.86 (br, 2H), 5.19 (s, 1H), 5.83 (t, J=10.0 Hz, 1H), 5.87 (d, J=9.6 Hz, 1H), 6.59 (t, J=11.2 Hz, 1H), 6.96 (d, J=11.6 Hz, 1H), 7.15 (s, 1H) and 9.19 (s, 1H). MS(FAB) m/z calcd. for C34H50N4O9 658.4; found (M+H+) 659.3.
17-(2′-(1′-Azaheterocyclohexyl-1′-)ethylamino)-17-demethoxygeldanamycin (2i)
Compound 2i was synthesized (yield 42%) as a purple solid (HPLC purity: 99.9%). [α]43620=+904.9° (c=0.0156, CH3OH). m.p. 211–214 °C. The 1H NMR (400 MHz, CDCl3): δ 0.85–0.98 (m, 6H), 1.01–1.36 (m, 4H), 1.48–1.51 (m, 3H), 1.62–1.70 (m, 5H), 1.81 (s, 3H), 2.03 (s, 3H), 2.32–2.37 (m, 4H), 2.42 (s, 1H), 2.59–2.67 (m, 4H), 2.74–2.86 (m, 1H), 3.27 (s, 3H), 3.37 (s, 3H), 3.43 (d, J=9.2 Hz, 1H), 3.58 (d, J=9.2 Hz, 1H), 3.68 (br, 1H), 4.31 (d, J=10.0 Hz, 1H), 4.50 (br, 1H), 4.78 (br, 2H), 5.19 (s, 1H), 5.83 (t, J=10.4 Hz, 1H), 5.93 (d, J=9.6 Hz, 1H), 6.56 (t, J=11.6 Hz, 1H), 6.97 (d, J=11.6 Hz, 1H), 7.25 (br, 1H) and 9.19 (s, 1H). MS(FAB) m/z calcd. for C35H52N4O8 656.4; found (M+) 656.3.
17-[(1′R,5′S)-6′,6′-dimethylbicyclo[3.1.1]heptan-3′-yl)methanamine]-17-demethoxygeldanamycin (2j)
Compound 2j was synthesized (yield 56%) as a purple solid (HPLC purity: 99.7%). [α]43620=+625.0° (c=0.0156, CH3OH). m.p. 130–133 °C. The 1H NMR (400 MHz, CDCl3): δ 0.87–0.98 (m, 6H), 1.02 (s, 3H), 1.31 (s, 3H), 1.55–1.60 (m, 3H), 1.72 (m, 1H), 1.80 (s,3H), 1.93–1.98 (m, 6H), 2.03 (s, 3H), 2.31–2.77 (m, 5H) 3.27 (s, 3H), 3.37 (s, 3H), 3.40–3.60 (m, 4H), 4.30 (d, J=10.0 Hz, 2H), 4.37 (br, 1H), 4.72 (br, 2H), 5.19 (s, 1H), 5.86 (t, J=11.2 Hz, 1H), 5.90 (d, J=10.0 Hz, 1H), 6.38 (br,1H), 6.58 (t, J=11.5 Hz, 1H), 6.95 (d, J=11.5 Hz, 1H) and 9.14 (s, 1H). MS(FAB) m/z calcd. for C38H55N3O8 681.4; found (M+) 681.3.
17-(3′-(2′-Amino-5′-nitropyridin) ethylamine)-17-demethoxygeldanamycin (2k)
Compound 2k was synthesized (yield 40%) as a purple solid (HPLC purity: 98.3%). [α]43620=+566.7° (c=0.0150, CH3OH). m.p. 131–134 °C. The 1H NMR (400 MHz, CDCl3): δ 0.94–1.01 (m, 6H), 1.26–1.28 (m, 2H), 1.52 (s, 1H), 1.68 (s, 2H), 1.80 (s, 3H), 2.02 (s, 3H),2.38–2.42 (m, 1H), 2.67–2.71 (m, 2H), 2.74–2.76 (m, 2H), 3.27 (s, 3H), 3.36 (s, 3H), 3.46–3.47 (m, 1H), 3.72 (d, 1H), 3.84 (br, 1H), 4.31 (d, J=10.0 Hz, 1H), 4.75 (br, 2H), 5.19 (s, 1H), 5.56 (br, 1H), 5.83–5.90 (m, 2H), 6.46 (br, 1H), 6.58 (t, J=11.2 Hz, 1H), 6.96 (t, J=11.2 Hz, 1H); 7.21 (s, 1H), 7.26–7.29 (m, 1H), 8.21 (s, 2H) and 9.12 (br, 1H). MS(ESI) m/z calcd. for C35H46N6O10 710.3; found (M+Na+) 733.4.
Tert-butyl-2-nicotinamido-ethylcarbamate (5)
Nicotinic acid 1.8 g (0.015 mol) was suspended in 5 ml of methylene dichloride. Thionyl chloride 4.4 ml was added. The mixture was refluxed under N2 for 5 h. After the white solid was transformed into an acicular crystal, the mixture was filtered, and the filter cake was washed with methylene dichloride and nicotinoyl chloride (4) was obtained.
The nicotinoyl chloride produced above, in 5 ml triethylamine, was added to a mixture of 2 ml dichloromethane and 2 ml tetrahydrofuran, containing N-Boc-ethylendiamine 0.16 g with stirring at room temperature under N2. The reaction solution was filtered after 2 h, concentrated in a rotatory evaporator and purified by flash chromatography on silica gel to give tert-butyl-2-nicotinamido ethylcarbamate (5) (yield 75%). The 1H NMR (400 MHz, CDCl3): δ 1.43–1.44 (m, 9H), 3.43 (m, 2H), 3.58 (m, 2H), 5.06 (br, 1H), 7.38–7.41 (m, 1H), 7.63 (br, 1H), 8.19 (d, J=7.6 Hz, 1H), 8.72 (d, J=7.6 Hz, 1H) and 9.09 (s, 1H).
2-Nicotinamido-ethylamino hydrochloride (6)
Compound 5 was suspended in 4 ml MeOH, and 2 ml acetyl chloride was added dropwise at <5 °C. Then the mixture was stirred for 1 h at room temperature and filtered. The filter cake was washed with EtOAc. The 2-nicotinamido-ethylamine hydrochloride (6) was obtained (yield 79%). The 1H NMR (400 MHz, CDCl3): δ 2.99 (m, 2H), 3.51–3.53 (m, 2H), 7.58 (d, 1H), 7.87 (br, 2H), 8.28 (s, 1H), 8.74 (d, J=7.6 Hz, 1H), 8.89 (br, 1H) and 9.06 (s, 1H).
17-(2′-Nicotinamidylethylamino)-17-demethoxygeldanamycin (2l)
To a suspension of GA (50 mg, 89.29 μmol) in 5 ml of 10:1 CHCl3/MeOH, 2-nicotinamido-ethylamino hydrochloride (6) (74 mg, 258 μmol) and 0.5 ml of triethylamine were added. The reaction mixture was stirred at room temperature for 3 days. The mixture was concentrated on a rotary evaporator, suspended in EtOAc, washed sequentially with water, saturated aqueous sodium bicarbonate, 1 M aqueous hydrochloric acid and saturated aqueous sodium chloride. The organic solution was evaporated to dryness, giving a purple solid. Purification was carried out by flash chromatography over silica gel (eluted using dichloromethane/MeOH (50/1-30/1)) to afford the title compound 2l (yield 79%) as a purple solid (HPLC purity: 99.0%). [α]43620=+469.3° (c=0.0160, CH3OH). m.p. 119–122 °C. The 1H NMR (400 MHz, CDCl3): δ 0.98–0.99 (m, 6H), 1.80 (s, 3H), 2.02 (s, 3H), 2.42–2.46 (m, 1H), 2.65 (d, J=14.0 Hz, 1H), 2.732–2.76 (m, 1H), 3.09–3.12 (m, 2H), 3.27 (s, 3H), 3.35 (s, 3H), 3.42–3.57 (m, 2H), 3.79–3.93 (m, 3H), 4.30 (d, J=10.0 Hz, 1H), 4.80 (br, 2H), 5.17 (s, 1H), 5.30 (br, 1H), 5.84–5.90 (m, 2H), 6.57 (t, J=11.5 Hz, 1H), 6.93–6.95 (m, 2H), 7.21 (s, 1H), 7.53 (s, 1H), 8.18 (s, 1H), 8.44 (d, J=7.5 Hz, 1H), 8.76 (s, 1H), 9.13 (br, 1H), 9.34 (s, 1H) and 11.89 (br, 3H). MS(FAB) m/z calcd. for C36H47N5O9 693.3; found (M+H+) 694.6.
17,19-di-(R)-THFM-17-demethoxygeldanamycin (3)
To a suspension of GA (500 mg, 892.9 μmol) in 5 ml of 10:1 CHCl3/MeOH, (R)-tetrahydrofurfurylamine (4.5 mmol) was added at room temperature. The reaction mixture was stirred for 2 days until the reaction was completed as monitored by TLC. The reaction work-up was the same as that described for the 17 position singly substituted compound 2b. The title compound 3 was obtained (yield 78%) as a purple solid (HPLC purity: 98.0%), [α]43620=+34.8° (c=0.0177, CH3OH). m.p. 92–95 °C. The 1H NMR (600 MHz, CDCl3): δ 0.78 (d, J=6.0 Hz, 3H), 1.03 (d, J=7.2 Hz, 3H), 1.07–1.11 (m, 1H), 1.53–1.78 (m, 8H), 1.83–2.50 (m, 12H), 2.28–2.51 (m, 3H), 3.08–3.17 (m, 2H), 3.20–3.27 (m, 1H), 3.28 (s, 3H), 3.31 (s, 3H), 3.47–3.55 (m, 1H),3.58–3.70 (m, 2H), 3.76–3.82 (m, 2H), 3.85–3.90 (m, 2H), 4.04–4.15 (m, 2H), 4.32–4.38 (dd, J1=8.4 Hz, J2=6.6 Hz, 1H), 4.88 (br, 2H), 5.07 (d, J=6.6 Hz, 1H), 5.26–5.31 (m, 2H), 5.57 (t, J=10.2 Hz, 1H), 6.53 (t, J=12.0 Hz, 1H) and 7.36 (d, J=12.0 Hz, 1H). HR-MS(ESI) m/z calcd. for C38H58N4O10Na 753.40506; found (M+Na+) 753.40417
Pharmacology
HCV replicon cell
FCA4(GS4.3), a human hepatoma HuH-7 cell line carrying an HCV subgenomic replicon I 377-3′del.S,15 was maintained in Dulbecco's modified Eagle's medium (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Gibco-BRL), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 500 μg ml−1 of Geneticin (G418; Invitrogen, Carlsbad, CA, USA) at 37 °C in 5% CO2. The HCV replicon was constructed as shown in Figure 1.
HCV RNA replication inhibitory assay
GS4.3 cell monolayers were digested with trypsin/EDTA and cell numbers were calculated. A volume of 100 μl GS4.3 cells (5 × 104 cells per ml) was placed on a 96-well plate. After 24 h, the supernatants were replaced with new culture media containing various concentrations of IFN-α-2b (Intron A; Schering Plough, Kenilworth, NJ, USA) or GA and its derivatives. At 72 h, the cells toxicity was detected using the tetrazolium (MTT) assay, and intracellular HCV RNA was quantified.
Synergistic inhibition on HCV RNA by 2b in combination with IFN-α-2b
After defining the optimum starting concentrations for each inhibitor, 2b was examined in combination with Intron A. Inhibition assays were set up as described above using GS4.3 as the target cells. The concentrations of each inhibitor were chosen so as to ensure a minimum of data points between 20 and 80% inhibition. The assay was then performed as described above.
Real-time PCR for detecting HCV RNA in replicon cells
Total intracellular RNA was extracted using the RNeasy mini kit (Qiagen, Hilden, Germany). The amount of RNA was estimated by SuperScript III Platinum SYBR Green One-Step qRT-PCR Kit (Invitrogen) according to the manufacturer's protocol. Fluorescent signals were analyzed by the iQ5 real-time PCR detection system (Bio-Rad laboratories, Madrid, Spain) using the primer pairs of 5′-CGGGAGAGCCATAGTGGTCTGCG-3′ and 5′-CTCGCAAGCACCCTATCAGGCAGTA-3′.16

The synthetic procedure for preparation of geldanamycin derivatives.

The synthetic procedure for 2l.
References
Koziel, M. J. & Peters, M. G. Viral hepatitis in HIV infection. N. Engl. J. Med. 356, 1445–1454 (2007).
Zeuzem, S. Interferon-based therapy for chronic hepatitis C: current and future perspectives. Nat Clin Pract Gastroenterol Hepatol. 5, 610–622 (2008).
Moriishi, K. & Matsuura, Y. Evaluation systems for anti-HCV drugs. Adv. Drug Deliv. Rev. 59, 1213–1221 (2007).
Pawlotsky, J. M., Chevaliez, S. & McHutchison, J. G. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology. 132, 1979–1998 (2007).
Kronenberger, B., Welsch, C., Forestier, N. & Zeuzem, S. Novel hepatitis C drugs in current trials. Clin Liver Dis. 12, 529–555 (2008).
Soriano, V., Peters, M. G. & Zeuzem, S. New therapies for hepatitis C virus infection. Clin. Infect. Dis. 48, 313–320 (2009).
Klebl, B. M., Kurtenbach, A., Salassidis, K., Daub, H. & Herget, T. Host cell targets in HCV therapy: novel strategy or proven practice? Antivir. Chem. Chemother. 16, 69–90 (2005).
Tai, A. W. et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 5, 298–307 (2009).
Moriishi, K. & Matsuura, Y. Host factors involved in the replication of hepatitis C virus. Rev. Med. Virol. 17, 343–354 (2007).
Okamoto, T. et al. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. Eur. Mol. Biol. Organ. 25, 5015–5025 (2006).
Nakagawa, S. I. et al. Hsp90 inhibitors suppress HCV replication in replicon cells and humanized liver mice. Biochem. Biophys. Res. Commun. 353, 882–888 (2007).
Schnur, R. C. et al. Inhibition of the oncogene product p185erbB−2in vitro and in vivo by geldanamycin and dihydrogeldanamycin derivatives. J. Med. Chem. 38, 3806–3812 (1995).
Lanford, R. E. et al. Antiviral effect and virus-host interactions in response to alpha interferon, gamma interferon, poly(I)-poly(C), tumor necrosis factor alpha, and ribavirin in hepatitis C virus subgenomic replicons. J. Virol. 77, 1092–1104 (2003).
Liu, W. L. et al. Ribavirin up-regulates the activity of double-stranded RNA-activated protein kinase and enhances the action of interferon-α against hepatitis C virus. J. Infect. Dis. 196, 425–434 (2007).
Lohmann, V. et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 285, 110–113 (1999).
Yang, J. H. et al. Real-time RT-PCR for quantitation of hepatitis C virus RNA. J. Virol. Methods. 102, 119–128 (2002).
Acknowledgements
This work was supported by the National Nature Science Foundation of China (Grant 30472076), ‘863’ program from the Ministry of Science and Technology of China (2006AA028414) and the National Major Science and Technology Project of China (‘Innovation and Development of New Drugs’, 2009ZX09301-003).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Shan, Gz., Peng, Zg., Li, Yh. et al. A novel class of geldanamycin derivatives as HCV replication inhibitors targeting on Hsp90: synthesis, structure–activity relationships and anti-HCV activity in GS4.3 replicon cells. J Antibiot 64, 177–182 (2011). https://doi.org/10.1038/ja.2010.161
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2010.161
Keywords
This article is cited by
-
Synthesis and biological evaluation of geldanamycin analogs against human cancer cells
Cancer Chemotherapy and Pharmacology (2015)
-
Geldanamycin, a ligand of heat shock protein 90, inhibits herpes simplex virus type 2 replication both in vitro and in vivo
The Journal of Antibiotics (2012)
-
Antiviral drugs against hepatitis C virus
Genetic Vaccines and Therapy (2011)
