
Abstract
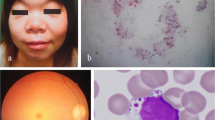
Two new individuals with α-NAGA deficiency are presented. The index patient, 3 years old, has congenital cataract, slight motor retardation and secondary demyelinisation. Screening of his sibs revealed an α-NAGA deficiency in his 7-year-old healthy brother who had no clinical or neurological symptoms. Both sibs are homozygous for the E325K mutation, the same genotype that was found in the most severe form of α-NAGA deficiency presenting as infantile neuroaxonal dystrophy. Thus, at the age of 7 years the same genotype of α-NAGA may present as a ‘non-disease’ (present healthy case) and can be associated with the vegetative state (the first two patients described with α-NAGA deficiency). The clinical heterogeneity among the 11 known individuals with α-NAGA deficiency is extreme, with a ‘non-disease’ (two cases) and infantile neuroaxonal dystrophy (two cases) at the opposite sides of the clinical spectrum. The broad spectrum is completed by a very heterogeneous group of patients with various degrees of epilepsy/behavioural difficulties/psychomotor retardation (four patients) and a mild phenotype in adults without overt neurological manifestations who have angiokeratoma and clear vacuolisation in various cell types (three cases). These observations are difficult to reconcile with a straightforward genotype-phenotype correlation and suggest that factors or genes other than α-NAGA contribute to the clinical heterogeneity of the 11 patients with α-NAGA deficiency.
Similar content being viewed by others


Article PDF
References
van Diggelen OP, Schindler D, Kleijer WJ et al:. Lysosomal α-N-acetylgalactosaminidase deficiency: a new inherited metabolic disease. The Lancet 1987 II: 804.
Schindler D, Bishop DF, Wolfe DE et al:. Neuroaxonal dystrophy due to lysosomal α-N-acetylgalactosaminidase deficiency. New Engl J Med 1989 320: 1735–1740.
Kanzaki T, Mizuno N, Yokota M, Matsumoto Y, Hirabayashi Y: . Novel lysosomal glycoaminoacid storage disease with angiokeratoma corporis diffusum. Lancet 1989 I: 875–876.
Kanzaki T, Michiko Y, Fumitoshi I, Hirabayashi Y, Wang AM, Desnick RJ: . Angiokeratoma corporis diffusum with glycopeptiduria due to deficient lysosomal α-N-acetylgalactosaminidase activity. Arch Dermatol 1993 129: 460–466.
Yokota M, Koji M, Yotsumoto S: . Histopathologic and ultrastructural studies of angiokeratoma corporis diffusum in Kanzaki disease. J Dermatol 1995 22: 10–18.
Chabas A, Coll MJ, Aparicio M, Rodriguez Diaz E: . Mild phenotypic expression of α-N-acetylgalactosaminidase deficiency in two adult siblings. J Inher Metab Dis 1994 17: 724–731.
de Jong J, van den Berg C, Wijburg H et al:. α-N-acetylgalactosaminidase deficiency with mild clinical manifestations and difficult biochemical diagnosis. J Ped 1994 125: 385–391.
Linden HU, Klein RA, Egge H, Peter-Katalinic J, Dabrowski J, Schindler D: . Isolation and structural characterisation of sialic acid-containing glycopeptides of the O-glycosidic type from the urine of two patients with an hereditary deficiency in α-N-acetylgalactosaminidase activity. Biol Chem Hoppe-Seyler 1989 370: 661–672.
Hirabayashi Y, Matsumoto Y, Matsumoto M et al:. Isolation and characterisation of major urinary amino acid O-glycosides and a dipeptide O-glycoside from a new lysosomal storage disorder (Kanzaki disease). J Biol Chem 1990 265: 1693–1701.
Keulemans JLM, Reuser AJJ, Kroos MA et al:. Human α-N-acetylgalactosaminidase (α-NAGA) deficiency: new mutations and the paradox between genotype and phenotype. J Med Genet 1996 33: 458–464.
van Diggelen OP, Schindler D, Willemsen RJ et al:. α-N-acetylgalactosaminidase deficiency, a new lysosomal storage disorder. J Inher Metab Dis 1988 11: 349–357.
Abeling NGGM, Rusch H, van Gennip AH: . Improved selectivity of urinary oligosaccharide screening using two one-dimensional TLC systems. J Inher Metab Dis 1996 19: 260–262.
Gay C, Maire I, Piraud M, Blanchon YC, Lauras B, Desnick RJ: . Another phenotype of Schindler disease or α-N-acetylgalactosaminidase deficiency. 2ème Congrès Scientifique VML sur les Maladies Lysosomales. Dijon, France, 22–24 January, 1998.
Wenger DA, Sujanski E, Fennessey PV, Thompson JN: . Human β-mannosidase deficiency. New Engl J Med 1986 315: 1201–1205.
Wang AM, Schindler D, Desnick RJ: . Schindler disease: the molecular lesion in the α-N-acetylgalactosaminidase gene that causes an infantile neuroaxonal dystrophy. J Clin Invest 1990 86: 1752–1756.
Simonaro L, Maire I, Gay C, Desnick RJ: . 1999www.mssm.edu/crc/mutations/schindler.html
Wang AM, Kanzaki T, Desnick RJ: . The molecular lesion in the α-N-acetylgalactosaminidase gene that causes angiokeratoma corporis diffusum with glycopeptiduria. J Clin Inv 1994 94: 839–845.
Yamauchi T, Hiraiwa M, Kobayashi H, Uda Y, Miyatake T, Tsuji S: . Molecular cloning of two species of cDNAs for human α-N-acetylgalactosaminidase and expression in mammalian cells. Biochem Biophys Res Comm 1990 170: 231–237.
Acknowledgements
We thank Professor Hans Galjaard for his continuing support, Tom de Vries Lentsch for photography and Henny Rusch for performing the oligosaccharide analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bakker, H., de Sonnaville, ML., Vreken, P. et al. Human α-N-acetylgalactosaminidase (α-NAGA) deficiency: no association with neuroaxonal dystrophy?. Eur J Hum Genet 9, 91–96 (2001). https://doi.org/10.1038/sj.ejhg.5200598
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200598
Keywords
This article is cited by
-
Targeted next-generation sequencing analysis in couples at increased risk for autosomal recessive disorders
Orphanet Journal of Rare Diseases (2018)
-
Structural and immunocytochemical studies on α-N-acetylgalactosaminidase deficiency (Schindler/Kanzaki disease)
Journal of Human Genetics (2004)
