
Abstract
We performed whole-genome sequencing (WGS) of a case of early-stage small-cell lung cancer (SCLC) to analyze the genomic features. WGS revealed a lot of single-nucleotide variations (SNVs), small insertion/deletions and chromosomal abnormality. Chromosomes 4p, 5q, 13q, 15q, 17p and 22q contained many block deletions. Especially, copy loss was observed in tumor suppressor genes RB1 and TP53, and copy gain in oncogene hTERT. Somatic mutations were found in TP53 and CREBBP. Novel nonsynonymous (ns) SNVs in C6ORF103 and SLC5A4 genes were also found. Sanger sequencing of the SLC5A4 gene in 23 independent SCLC samples showed another nsSNV in the SLC5A4 gene, indicating that nsSNVs in the SLC5A4 gene are recurrent in SCLC. WGS of an early-stage SCLC identified novel recurrent mutations and validated known variations, including copy number variations. These findings provide insight into the genomic landscape contributing to SCLC development.
Similar content being viewed by others

Introduction
Small-cell lung cancer (SCLC) is characterized by rapid proliferation and early development of widespread dissemination. Thus, most patients were diagnosed with advanced-staged diseases. Although response rates to the initial therapy are impressive, most patients experience relapse within the first 2 years and die from systemic metastasis.1 Despite the collaborative efforts to improve treatment, survival from SCLC has not changed over the past 25 years.2 Thus, a more comprehensive understating of SCLC biology is needed to develop more effective treatment for this devastating disease. However, the lack of availability of surgical materials frequently hampers the study of SCLC biology. Recent advances in next-generation sequencing technologies provide a means of discovering mutational processes across the whole genome.3 Although a study has provided a comprehensive genomic view of SCLC using massively parallel sequencing, it sequenced an SCLC cell line derived from a bone marrow metastasis of an SCLC patient.3 Other recently published studies reported novel somatic driver mutations of SCLC by integrated analyses of various data sets generated by the next-generation sequencing method. They reported frequent inactivation of TP53, RB1 and PTEN in more than 100 samples, and oncogenic amplification of c-MYC, FGFR1 and SOX2.4, 5
Here, we analyzed whole genomes of matched normal tumor samples from a patient who underwent curative resection for stage IA SCLC in National Cancer Center, Korea. This paired normal tumor comparison could identify recurrent mutations when compared with previously known mutation profiles in SCLC. In other words, the complexity of SCLC and ethnic differences of somatic mutations for lung cancer propose the possibility of other variations associated with SCLC in the Korean ethnic group.6 The importance of early detection of SCLC was another point of focus of our study on early-stage SCLC. The data reported here increase our understanding of the pathogenesis of SCLC and will allow the development of more targeted therapies for SCLC.
Materials and methods
Patient and specimen collection
Tumor and normal tissue samples were obtained from a patient with SCLC who underwent curative resection. After the pathological examination, the tumor and normal tissue samples were snap frozen and maintained in liquid nitrogen until genomic DNA extraction. This study was conducted under the approval of the ethical review boards and as per the guidelines for good clinical practice. The study subject gave informed consent for the genomic analysis.
Genomic DNA preparation
The frozen tumor sample was microdissected and lightly stained with hematoxylin to identify the portion consisting of 80% or more cancer cells. The genomic DNA was extracted with a MagAttract DNA Blood Midi Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocols. The DNA quality was assessed with the use of an F200 spectrophotometer (Tecan, Männedorf, Switzerland). A260/280 value greater than 1.7 was accepted for further analysis. The DNA quantity was assessed with the use of a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA). Control DNA from matched normal tissue was processed in the same manner. The same frozen tumor samples were used for total RNA extraction using a QIGEN RNeasy Mini Kit (Qiagen). Quality of total RNA was assessed with lab-on-a-chip on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Whole-genome sequencing
A volume of 5 μg of genomic DNA was sheared using Covaris S series (Covaris, Woburn, MA, USA). The fragment of sheared DNA was end-repaired, A-tailed and ligated to paired-end adapters according to the manufacturer’s protocol (Pair End Library Preparation Kit, Illumina, San Diego, CA, USA). Adapter ligated fragments were purified and dissolved in 30 μl of elution buffer, and 1 μl of the mixture was used as a template for 12 cycles of PCR amplification. The PCR product was gel purified using a QIAquick Gel Extraction Kit (Qiagen). Library quality and concentration was determined using the Agilent 2100 BioAnalyzer (Agilent). Libraries were quantified using an SYBR green qPCR protocol on LightCycler 480 (Roche, Indianapolis, IN, USA) according to Illumina’s library quantification protocol. On the basis of the qPCR quantification, libraries were normalized to 2 nM and then denatured using 0.1 N NaOH. Cluster amplification of denatured templates occurred in flow cells, according to the manufacturer’s protocol (Illumina). Flow cells were paired‐end sequenced (2 × 100 bp) on an Illumina HiSeq 2000 using HiSeq Sequencing kits. A base-calling pipeline (Sequencing Control Software, SCS; Illumina) was used to process the raw fluorescent images and the called sequences.
Reads alignment and variation detection
Ninety-base-pair paired-end sequence reads with ∼300 bp insert size were aligned to hg19 human reference genome (NCBI build 37) with a BWA algorithm ver. 0.5.9.7 Two mismatches were permitted in the 45 bp seed sequence. To remove PCR duplicates of sequence reads, which can be generated during the library construction process, we used the ‘rmdup’ command of Samtools.8 Aligned reads were realigned at putative indel positions with the GATK IndelRealigner algorithm to enhance mapping quality. Base quality scores were recalibrated using the TableRecalibration algorithm of GATK.9
SNP and small insertion/deletion (Indel) analysis
Putative single-nucleotide variations (SNVs) were called and filtered using UnifiedGenotyper and VariantFiltration commands in GATK.9 The options used for SNP calling were a minimum of 5 to a maximum of 200 read mapping depth with consensus quality 20, and the prior likelihood for heterozygosity value 0.001. To obtain somatic mutations in cancer genomes, SNVs from cancer genomes were filtered using the SNVs from normal tissue genomes. The remaining SNVs were filtered again using the mapping status of normal tissue genomes. At each remaining tumor SNV position, if the minimum mapping depth was at least 3 and the SNV nucleotide ratio was at least 0.2 in normal tissue genomes, the tumor SNV was discarded. To obtain somatic small indels, the IndelGenotyperV2 paired-sample mode of GATK was used.9 Window size 300 and other default options were used. All somatic mutations altering amino acid were checked by expert lab personnel using the tview command of Samtools.8 If an SNV was in a low-quality region or germline mutation, it was discarded. Twenty-one novel indels in frameshift were validated using Samtools Tview with depth <5, depth rate <0.3, continuous mapped region <2 in both 10 bp sides and continuous mapped region rate <0.1 to remove false-positive results (Supplementary Table 5).
Annotation of variations
Predicted SNVs were compared with NCBI dbSNP version 131 (http://www.ncbi.nlm.nih.gov/projects/SNP/) to annotate known SNP information.10 Each SNV was mapped on the genomic features of the UCSC gene table, such as coding region, untranslational region and intron. Nonsynonymous (ns) SNV information was extracted by comparing UCSC (http://genome.ucsc.edu/) reference gene information. KEGG (http://www.genome.jp/kegg/) and Biocarta (http://www.biocarta.com/) pathways were used to analyze altered protein sets. Information on cancer-related mutations was obtained from the cosmic cancer information database (http://www.sanger.ac.uk/genetics/CGP/cosmic/).
Identification of copy number variation regions
Owing to the heterogeneity of cancer samples, a new method was developed for identifying copy number variations based on the differences of sequencing depths between normal and cancer samples. The program defined the CNV regions containing the borders that presented significant differences by considering each pair of samples. Final CNV regions were defined by merging adjacent CNV regions with similar copy numbers. To calculate the frequencies of duplication or deletion events, the number of CNV regions in tumor samples that showed duplication or deletion was counted for all 23 chromosomes.
Genome-wide SNP analysis
SNP genotyping was performed using an Axiom genotyping solution including an Axiom Genome-Wide ASI 1 Array Plate and reagent kit according to the manufacturer’s protocol (Affymetrix, Santa Clara, CA, USA). Briefly, total genomic DNA (200 ng) was treated with 20 μl of denaturation buffer and 40 μl of neutralization buffer, followed by amplification for 23 h using 320 μl of Axiom amplification mix. Amplified DNA was randomly fragmented into 25–125 bp size with 57 μl of Axiom fragmentation mix at 37 °C for 30 min, followed by DNA precipitation for DNA clean-up and recovery. DNA pellets were dried and resuspended with 80 μl of hybridization master mix. A volume of 3 μl of suspended sample was kept for sample qualification. A hybridization ready sample was denaturated using a PCR machine at 95 °C for 20 min and 48 °C for 3 min. Denatured DNA was transferred to a hybridization tray and loaded to a GeneTitan MC with Axiom ASI array plate (Affymetrix). Hybridization continued on the GeneTitan for 24 h, followed by loading ligation, staining and stabilization reagent trays into the instrument. GeneTitan was controlled by an Affymetrix GeneChip Command Console GeneTitan Control (Affymetrix). The chip image was scanned with the GeneTitan and the resulting data, in a dat file, were automatically transformed to a cel file as a final intensity file. To genotype call, the cel intensity file was normalized, and genotype calling was done using Genotyping Console 4.1 with Axiom GT1 algorithms according to the manufacturer’s manual. The cutoff values for data quality control were DISHQC ⩾0.82 for hybridization, and the QC call rate was ⩾97%.
Validation of SNVs by Sanger sequencing
SNVs were validated by conventional Sanger sequencing using dye-terminator chemistry analyzed with an automatic sequencer ABI 3730 (Applied Biosystems, San Diego, CA, USA). The target regions were amplified by PCR followed by direct sequencing, or cloned into TA vectors. At least 20 TA vector clones were sequenced, because mutations with low purity are difficult to detect by Sanger sequencing.
Structural variants (SVs) and gene fusion analysis
SVs are analyzed using breakdancer.11 An SV not found in normal tissue samples is defined as somatic SV. Gene fusions are analyzed using SVs by defining fusion signals that inform fusion point and fusion direction. We excluded fusion signals that were found not only from the same mate normal tissue but also from other 19 samples. We decided two fusion signals were equal if the distance between breakpoints was less than 1 kbp. When we scanned the gene fusion event, fusion signals not located in two genes and gene fusions causing opposite transcription of a constitute gene were excluded. Genes having SV breakpoints suffer gene breakage. When defining the gene breakage, SVs located in an intron were also excluded.
Results
A 57-year-old Korean man with former smoking history (average 20 cigarettes per day for 15 years) presented at our hospital with asymptomatic early-stage lung cancer detected by screening in June 2006. Ultimately, stage IA (by AJCC, the 6th edition) SCLC of the right upper lobe without regional lymph node involvement was diagnosed. He was treated with right upper lobe lobectomy and the final pathologic stage was T2N0M0. He received four cycles of adjuvant chemotherapy with irinotecan and cisplatin after surgery. At present, the patient is alive without other recurrence of his disease (January 2013).
On average, 99 gigabases per sample were produced at ∼33X sequencing depth, and they were mapped to the reference genome (NCBI build 37, HG19) at an over 95% mapping rate (Supplementary Table 1). Among the mapped reads, the reads inside the mean/standard deviation range (properly mapped reads) were over 93% and the reads that did not make it into the contigs (singletons) were less than 5%. Therefore, we got qualified sequencing reads to cover the whole genome. Using the final properly mapped reads, we constructed a genomic profile database for detecting single nucleotide variations (SNVs), copy number variations (CNVs), structural variations (SVs) and fusion genes.
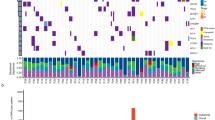
Structural variation analysis showed that genome duplications and deletions occurred randomly (Figure 1a). In the tumor genome, there are 465 large deletions, 23 medium-sized insertions, 15 inversions, 39 intra-chromosomal translocations and 18 inter-chromosomal translocations of breakdancer score ⩾80 (Supplementary Table 2). Overall, deletion events occurred 20 times more in length than in duplication, suggesting that genome-wide damage by deletion has a higher impact than amplification in SCLC. The structural variants in genomic regions were further analyzed by comparing with the cancer-related genes in the COSMIC (Catalog Of Somatic Mutations In Cancer, http://www.sanger.ac.uk/genetics/CGP/cosmic/) database. Six genes showed more than 300 bp of large structural variations, including four deletions and two inversions (Supplementary Table 3). The copy number variation (CNV) analysis revealed that chromosomes 4p, 5q, 13q, 15q, 17p and 22q contained many blocks of deletion. In contrast, chromosome 5p showed notably increased duplicated blocks (Figure 1a). When the exact loci of tumor suppressors and oncogenes were analyzed, we found that tumor suppressors RB1 at chromosome 13q and TP53 at chromosome 17p were in highly deleted chromosomal regions. In contrast, the copy number of oncogene hTERT at chromosome 5p was increased.

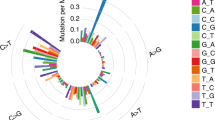
(a) Variations of the tumor genome. From the outer side of each ring, chromosome numbers and mapping depths of chromosome regions are indicated by numbers. Large translocations of intra- and inter-chromosomal rearrangements are indicated by color lines across the center. (b) Single-nucleotide substitution pattern in somatic mutations.
We also identified about 0.5 million short insertions and deletions (indel) in normal and tumor tissues. By subtracting indels found in the normal genome, 6 430 somatic indels specific for SCLC were identified (Supplementary Table 4). Among them, 21 novel indels from 21 genes resulted in frameshifts (Supplementary Table 5), which most likely damage the protein function. Interestingly, they are not previously reported as cancer-related genes.
By sequencing normal and tumor genomes, we identified 3.6 million SNVs. The results of NGS were also examined with Axiom genome-wide genotyping microarrays (Affymetrix). The genotyping data from microarrays showed 99.8% of concordance with the NGS data. By subtracting SNVs found in the normal genome, we were able to identify 62 763 somatic SNVs specific for SCLC (Table 1). Among them, there were 43 339 novel SNVs including 116 nsSNVs from 108 genes. A part of nsSNVs were validated and confirmed by conventional Sanger sequencing (Table 2). In addition, we analyzed the pattern of single-nucleotide substitutions in total somatic SNVs. The result shows that G>A/C>T (29%) and A>G/T>C (24%) transitions were more common than G>T/C>A (19%) transversions (Figure 1b).
In detail, we found a somatic mutation in the TP53 gene, which resulted in H193R amino acid change (Figure 2a). Together with the identification of loss of heterozygosity (LOH) in chromosomal regions covering the TP53 gene, the defected function of TP53 may be involved in carcinogenesis of this patient. A heterozygous mutation in the CREBBP gene was also found. This mutation changed Tyrosine at the 1395 amino acid position to Cysteine (Y1395C), which could affect the function of CREBBP transcription factor by interrupting the HAT domain (Figure 2b). Many other nsSNVs were found in a number of genes, which are not well analyzed functionally in cancer. For example, we identified a somatic nsSNV in the C6orf103 (Calpain-7 like protein) gene, which was also reported by whole-genome sequencing of an SCLC cell line.3 This mutation changed Valine at the 457 amino acid position to Methionine (Figure 2c). A novel nsSNV was also found in the SLC5A4 gene, which encodes a family member of low-affinity sodium-glucose cotransporters. This mutation changed Phenylalanine at the 17 amino acid position to Histidine (Figure 2d). One more nsSNV changing Glycine at the 43 amino acid position to Glutamic acid was observed by analyzing the exonic regions of the SLC5A4 gene in 23 additional SCLC samples, showing together 8.3% (2/24) frequency of nsSNV in the SLC5A4 gene.

Schematic of TP53 (a), CREBBP (b), C6ORF103 (c), SLC5A4 (d) protein illustrating functional domains with the location of their mutations.
Discussion
As SCLC is characterized by rapid proliferation and early dissemination, most cases present with advanced-stage diseases, which hamper the detection of early-stage SCLC. In this study, we analyzed the genomic profile of early-stage SCLC by whole-genome sequencing of matched normal tumor samples from a patient with stage IA SCLC who underwent curative resection. We identified 43 339 novel somatic SNVs and found their base substitution pattern different from that of heavy smokers. Many somatic SNVs and copy number variations have been already reported as either oncogenes or tumor suppressors in the COSMIC database, including TP53 and RB1 genes. This cancer genome had few mutated genes in the SCLC pathway, but showed statistically meaningful genetic changes in the Notch and WNT signaling pathways. Taken together, a comprehensive analysis of the whole genome from a patient with early-stage SCLC provided a distinct genomic profile, which may give insight into the molecular classification of early-stage SCLC patients for personalized diagnostics and treatment.
The mutation patterns in this study are somewhat different from that of heavy-smoking-related lung cancer. Only three types of mutations showed consistent differences in relation to tobacco smoking.3, 12 Both G>T/C>A transversions and A>G/T>C transitions were elevated in ever smokers compared with never smokers, whereas G>A/C>T transitions are decreased in a progressive manner with cumulative exposure to tobacco. In our study, the patient was a former smoker with a 15 pack-year smoking history who quit smoking 6 years ago before the diagnosis of SCLC. Thus, the relatively higher frequency of G>A/C>T transitions (29%) compared with G>T/C>A transversions (19%) in b could be caused by the mild smoking habit of the patient.
SCLC cells commonly contain somatic mutations in tumor suppressor genes, including TP53, RB1, p16, RASSF1A, CREBBP and FHIT genes.13 In this study, we found variations in TP53, RB1 and CREBBP genes, but not in p16, RASSF1A and FHIT genes. In particular, a TP53 mutation resulted in an H193R amino acid change, which resides in the DNA binding domain of TP53. It was reported that the TP53 H193R mutant protein weakly binds to and transactivates the p21 gene, which results in abnormal regulation of DNA synthesis, cell cycle and apoptosis.14, 15, 16 Loss of TP53 and RB1 function possibly triggers cell-type-specific carcinogenesis in SCLC.17, 18 Together with the identification of loss of heterozygosity (LOH) in chromosomal regions covering TP53 and RB1 genes, possible loss of TP53 function by the H193R mutation may be involved in the early stages of carcinogenesis of SCLC. Another gene harboring the driver mutation is CREBBP, a transcriptional cofactor that acetylates proteins including TP53.19 The dominant mutations of the CREBBP gene were known to cause Rubinstein-Taybi syndrome and increased sensitivity to tumorigenesis,20, 21 as well as gene dosage-dependent embryonic development and proliferation.22 Truncated CREBBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism.23 In addition, the cancer genome of the NCI-H209 cell line harbors the fusion of the first two exons in the CREBBP gene to the BTBD12 gene and a homozygous CREBBP deletion was observed in two SCLC cell lines.3, 24 As a recent SCLC genome and transcriptome study reported that somatic mutations in CREBBP were clustered around the HAT domain encoding sequence,4 we also found an nsSNV at the HAT domain as a heterozygous mutation in CREBBP. The coexistance of TP53 and CREBBP mutations suggests that somatic mutations in those genes may affect multiple cancer-related pathways, as TP53 and CREBBP have overlapping roles in several different pathways.
Notably increased duplication of hTERT was observed is this study. It is reported that telomerase gene amplification appears to increase both hTERT mRNA expression and telomerase activity, which are necessary during the early phase of carcinogenesis. Increased hTERT mRNA and telomerase activity are frequently observed in SCLC.25 These finding suggest that increased telomerase activity by hTERT amplification may occur early in tumorigenic transformation and initiate SCLC. In addition to oncogenic amplification of c-MYC, FGFR1 and SOX2 in other massive SCLC genome sequencing studies,4, 5 our finding could consider hTERT amplification as a SCLC oncogenic pathway.
In addition to SNVs in hot target genes, we found two novel recurrent nsSNVs. Somatic nsSNVs in the C6orf103 gene were recurrent as reported by whole-exome sequencing of SCLC cell lines (H209, H289, H1450, HCC33 cells) and an SCLC patient tissue. They observed various somatic amino acid changes from the mutations (C181F, A304G, V457M, K650N, Y957H), suggesting that C6orf103 may be a novel tumor-related gene in SCLC.4, 5 C6orf103 (Calpain-7 like protein) is a member of the calpain protein family, which is a conserved family of cysteine proteinases that catalyze the controlled proteolysis of many specific substrates. Calpain activity is implicated in several fundamental physiological processes, including cytoskeletal remodeling, cellular signaling, apoptosis and cell survival.26 Calpain expression is increased in tumor cells.27, 28 Calpains are important for proteolysis of numerous substrates in tumor pathogenesis, such as inhibitors of nuclear factor-κB (IκB), focal adhesion kinases and talins and c-MYC.29, 30, 31, 32 Therefore, the C6orf103 protein could be interpreted as a tumor suppressor.
A novel nsSNV in the SLC5A4 gene was found together with a copy loss of the gene. One more nsSNV was observed by analyzing the exonic regions of the SLC5A4 gene in 23 SCLC samples. Therefore, 8.3% of the SCLC patients (2/24) showed an nsSNV in the SLC5A4 gene. As far as we know, a somatic nsSNV in the SLC5A4 gene was first reported by sequencing of an SCLC cell line (H1672 cells) and two SCLC patients by two different groups.4, 5 We also found recurrent SLC5A4 mutations from SCLC patients in this study, which are different from previous findings. SLC5A4 is a unique member of low-affinity sodium-glucose cotransporters (SLC5 family). Among glucose transporters, SLC5A4 cannot transport sugar, but acts as a sugar sensor.33 Therefore, damaging changes in SLC5A4 may result in the loss of glucose sensing, which is essential for the control of cell growth and proliferation. As cancer cells require energy to feed uncontrolled proliferation, they normally overexpress the glucose transporter family members. Therefore, loss-of-function variations in the glucose sensor may contribute to lack of negative feedback regulation of glucose transporter gene expression in cancer cells. Our finding could point toward a novel cancer pathway for SCLC tumorigenesis.
In summary, our study represents a comprehensive genomic profile of an early-stage SCLC. Despite the limitation in the sample set, our study may provide considerable understanding of early development of SCLC.
References
Masters GA, Declerck L, Blanke C, Sandler A, DeVore R, Miller K et al. Phase II trial of gemcitabine in refractory or relapsed small-cell lung cancer: Eastern Cooperative Oncology Group Trial 1597. J Clin Oncol 2003; 21: 1550–1555.
Lally BE, Urbanic JJ, Blackstock AW, Miller AA, Perry MC . Small cell lung cancer: have we made any progress over the last 25 years? Oncologist 2007; 12: 1096–1104.
Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010; 463: 184–190.
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012; 44: 1104–1110.
Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 2012; 44: 1111–1116.
Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res 2012; 22: 2109–2119.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2008; 36 (Database issue): D13–D21.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 2009; 6: 677–681.
Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012; 150: 1121–1134.
Sanchez-Cespedes M . Lung cancer biology: a genetic and genomic perspective. Clin Transl Oncol 2009; 11: 263–269.
Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ . The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993; 75: 805–816.
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–825.
Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR . Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res 1994; 211: 90–98.
Sozzi G, Miozzo M, Pastorino U, Pilotti S, Donghi R, Giarola M et al. Genetic evidence for an independent origin of multiple preneoplastic and neoplastic lung lesions. Cancer Res 1995; 55: 135–140.
Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A . Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011; 19: 754–764.
Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG . Roles of CREB-binding protein (CBP)/p300 in respiratory epithelium tumorigenesis. Cell Res 2007; 17: 324–332.
Janknecht R . The versatile functions of the transcriptional coactivators p300 and CBP and their roles in disease. Histol Histopathol 2002; 17: 657–668.
Miller RW, Rubinstein JH . Tumors in Rubinstein-Taybi syndrome. Am J Med Genet 1995; 56: 112–115.
Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D et al. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998; 93: 361–372.
Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M et al. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Mol Genet 1999; 8: 387–396.
Kishimoto M, Kohno T, Okudela K, Otsuka A, Sasaki H, Tanabe C et al. Mutations and deletions of the CBP gene in human lung cancer. Clin Cancer Res 2005; 11 (2 Pt 1): 512–519.
Nishio Y, Nakanishi K, Ozeki Y, Jiang SX, Kameya T, Hebisawa A et al. Telomere length, telomerase activity, and expressions of human telomerase mRNA component (hTERC) and human telomerase reverse transcriptase (hTERT) mRNA in pulmonary neuroendocrine tumors. Jpn J Clin Oncol 2007; 37: 16–22.
Suzuki K, Hata S, Kawabata Y, Sorimachi H . Structure, activation, and biology of calpain. Diabetes 2004; 53 (Suppl 1): S12–S18.
Lee SJ, Choi YL, Lee EJ, Kim BG, Bae DS, Ahn GH et al. Increased expression of calpain 6 in uterine sarcomas and carcinosarcomas: an immunohistochemical analysis. Int J Gynecol Cancer 2007; 17: 248–253.
Lee SJ, Kim BG, Choi YL, Lee JW . Increased expression of calpain 6 during the progression of uterine cervical neoplasia: immunohistochemical analysis. Oncol Rep 2008; 19: 859–863.
Chan KT, Bennin DA, Huttenlocher A . Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J Biol Chem 2010; 285: 11418–11426.
Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR et al. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol 2004; 6: 977–983.
Han Y, Weinman S, Boldogh I, Walker RK, Brasier AR . Tumor necrosis factor-alpha-inducible IkappaBalpha proteolysis mediated by cytosolic m-calpain. A mechanism parallel to the ubiquitin-proteasome pathway for nuclear factor-kappab activation. J Biol Chem 1999; 274: 787–794.
Small GW, Chou TY, Dang CV, Orlowski RZ . Evidence for involvement of calpain in c-Myc proteolysis in vivo. Arch Biochem Biophys 2002; 400: 151–161.
Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C et al. A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci USA 2003; 100: 11753–11758.
Acknowledgements
This study was supported by a grant (No. 1110100) from the National Cancer Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website
Supplementary information
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Han, JY., Lee, YS., Kim, B. et al. Whole-genome analysis of a patient with early-stage small-cell lung cancer. Pharmacogenomics J 14, 503–508 (2014). https://doi.org/10.1038/tpj.2014.17
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2014.17
