
Abstract
Expression quantitative trait loci (eQTL) analysis is a powerful approach toward identifying genetic loci associated with quantitative changes in gene expression. We applied genome-wide association analysis to a data set of >300 000 single-nucleotide polymorphisms and >48 000 mRNA expression phenotypes obtained by Illumina microarray profiling of 149 human surgical liver samples obtained from Caucasian donors with detailed medical documentation. Of 1226 significant associations, only 200 were validated when comparing with a previously published similar study. Potential explanations for low replication rate include differences in microarray platforms, statistical modeling, covariates considered and origin and collection procedures of tissues. Focused analysis revealed a subset of 95 associations related to absorption, distribution, metabolism and excretion of drugs. Of these, 21 were true replications and 74 were newly discovered associations in enzymes, transporters, transcriptional regulators and other genes. This study extends our knowledge about the genetics of inter-individual variability of gene expression with particular emphasis on pharmacogenomics.
Similar content being viewed by others


Introduction
Genetic variants can affect qualitative and quantitative aspects at all levels of gene expression, including gene transcription, splicing, transcript stability, rate of translation, protein function and degradation, thereby contributing to inter-subject variability and heritable metabolic, pharmacogenetic and other phenotypes. Many variants, in particular, common single-nucleotide polymorphisms (SNPs), affect gene expression in a quantitative manner, and the combination of larger sets of low-impact variants is believed to explain non-Mendelian types of inheritance, including complex quantitative traits such as body size.1, 2, 3 Typical pharmacological phenotypes, such as drug response and toxicity, are highly likely to depend on multiple genes. In contrast to monogenically inherited pharmacogenetic polymorphisms, most of which have been discovered by following up on unusual clinical drug response phenotypes,4 the basis for more complex phenotypes remained largely unknown.5, 6
A relatively new approach to identify unknown functional genetic variants that modulate gene expression, also termed ‘genetical genomics,’ is the mapping of expression quantitative trait loci (eQTLs) using genome-wide association (GWA) methods in cohorts of unrelated individuals.2, 7 In this strategy, individual transcript levels are determined in a selected tissue or cell type using microarrays. In genomic DNA of the same individuals, in the order of 105 to 106 SNPs are genotyped in parallel. By considering each individual gene transcript as a quantitative trait, association analysis identifies SNPs that are significantly associated with expression.8, 9 Thus, the eQTL strategy differs from the typical GWA studies, as the majority of the >1000 published GWA studies typically focused on a single or only a few complex phenotypes.10
So far, only a limited number of genome-wide eQTL studies have been performed on various human tissues.11, 12, 13, 14, 15, 16, 17, 18, 19 In most cases, easily accessible peripheral tissues such as human HapMap lymphoblastoid cell lines, lymphocytes or monocytes were investigated. For example, in one of the earliest studies, Morley et al.20 distinguished cis- and trans-effects, depending on the relative location of trait gene and SNP gene to each other. Several later studies found that trans-eQTLs were more difficult to reproduce.12, 13, 14, 15 Only few studies have appeared on internal tissues, including the brain,21 adipose18 and liver.16 The latter study investigated a cohort of 427 human liver samples (in this paper referred to as the ‘Seattle study’) and found a multitude of new eQTLs. Furthermore, they showed that the eQTL approach together with network analyses can drive the identification of new susceptibility gene loci for complex disease traits such as type 1 diabetes.16
In this study, we investigated 149 human livers surgically removed from Caucasian donors to identify statistically significant associations between genetic polymorphisms and mRNA expression levels at a genome-wide scale. Although our study was similar in design and technology to the former study,16 the set of human liver samples had no overlap and differed in many aspects including ethnicity, sampling procedures, availability and completeness of clinical data. We focus in this paper on genes involved in absorption, distribution, metabolism and excretion (ADME) of drugs to allow for more detailed analyses, which resulted in a smaller set (∼20%) of truly replicated eQTLs and a larger set (∼80%) of unique eQTLs. This demonstrated that the genetical genomics approach is useful to identify novel genotype–phenotype relationships, and that a single study is insufficient to uncover all existing eQTLs in a given tissue.
Materials and methods
Liver samples
Liver tissues and corresponding blood samples were previously collected from 150 patients of Caucasian ethnicity (71 males and 79 females) undergoing liver surgery at the Campus Virchow (University Medical Center Charité, Humboldt University, Berlin, Germany). The average age of the subjects was 58±14 years. This study was approved by the ethics committees of the medical faculties of the Charité, Humboldt University, and of the University of Tuebingen and conducted in accordance with the Declaration of Helsinki. All tissue samples were examined by a pathologist and only histologically non-tumorous tissues were used. Clinical patient documentation available for all samples and shown to have significant influence on the analysis included age, sex, medical diagnosis (primary or secondary liver tumor, other diagnosis), presurgical medication (regular drug treatment before surgery vs no drugs), cholestatic liver injury (based on liver function tests22) and alcohol drinking and smoking habits. Patients with hepatitis, cirrhosis or chronic alcohol use were excluded. Detailed information on sample metadata is given in Supplementary Table S1.
Transcriptome analysis and genome-wide genotyping
RNA isolation from liver tissues was performed using Trizol (Invitrogen, Paisley, UK) extraction and Qiagen RNeasy-mini kit (Qiagen, Valencia, CA, USA) with on-column DNase treatment as described previously.23 Only high-quality RNA preparations according to Agilent Bioanalyzer (Nano-Lab Chip Kit, Agilent Technologies, Waldbronn, Germany) RNA Integrity Number (RIN) assignment (>7) were used in this study. In all, 200 ng of total RNA was amplified and labeled using the Illumina TotalPrep RNA amplification kit (Ambion Applied Biosystems, Darmstadt, Germany). cRNA quality was assessed by capillary electrophoresis on Agilent 2 100 Bioanalyzer (Agilent Technologies). Expression levels of >48 000 mRNA transcripts were assessed by Human-WG6v2 Expression BeadChip (Illumina, Eindhoven, The Netherlands). Hybridization was carried out according to the manufacturer's instructions. Genome-wide SNP data had been generated from genomic DNA using the HumanHap300 Genotyping BeadChip (Illumina) with 318 237 SNPs as described before.24 A comparison with the microarray platforms used in the Seattle study is shown in Figure 1. All data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE32504 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32504).

Distribution of SNPs among different genotyping platforms used in the Seattle and Stuttgart study. SNP, single-nucleotide polymorphism.
Preprocessing and quality control
Illumina BeadStudio version 3.0 (Illumina, San Diego, CA, USA) was used for all low-level preprocessing steps of the expression data, including background estimation and correction, normalization and probe set summary. After these low-level preprocessing steps, 9875 genes with high detection P-value (>0.1) or >10% missing values were filtered out and removed from the data set.25 The remaining missing signal intensities were estimated using the ‘k nearest neighbor’ algorithm implemented in R BioConductor.26, 27 The resulting data set was subsequently log2 transformed. Finally, after all preprocessing steps, the raw data of 48 701 probe signal intensities were mapped and reduced to signal intensities corresponding to 15 439 unique genes.
Raw data preprocessing of HumanHap300 Genotyping BeadChip was also performed using BeadStudio version 3.0. Next, missing genotypes were estimated using the MACH imputation algorithm, which is based on a hidden Markov model.28 Subsequently, 15 235 SNPs with an extremely low call rate (<95%),29 3466 SNPs with low minor allele frequencies (<4%)16 and 201 SNPs not in the Hardy–Weinberg equilibrium (false discovery rate ⩽0.2), were excluded from further analyses.16, 30 Genetic similarity between samples, referred to as population substructure, may lead to false-positive association results.31 To identify possibly related individuals, we calculated pairwise identity-by-state distances. Consequently, one sample was excluded because of >95% genotype identity to another sample.32 To detect further putative population substructures, the method of Price et al.33 based on principal components analysis (PCA) was applied. This analysis revealed no evidence for population substructure within our cohort of liver samples. This comprehensive quality control analysis was performed using the R BioConductor package ‘GenABEL’.34 The finally processed data set was from 149 livers (71 males and 78 females, Supplementary Table S1) and consisted of 299 352 SNPs and 15 439 gene expression levels. As a further internal control, we performed genotyping of sex chromosome-specific amelogenin gene variants35 and analysis of sex-specific gene expression (for example, XIST, RPS4Y, SMCY), which revealed 100% agreement with patient documentation.
GWA analysis
GenABEL34 was used to test all 4.6 billion possible combinations of SNPs and expression traits (299 352 SNPs*15 439 traits) for significant associations. Here, we assumed a genetic model in which both alleles contribute to gene expression in an additive manner, because this has been shown to be one of the most powerful statistical approaches.36 The SNP-trait associations were adjusted for sex, age, smoking, alcohol consumption, diagnosis, C-reactive protein level, cholestatic liver disease and presurgical medication as covariates (Supplementary Table S1), using an additive linear model.
Bonferroni’s multiple testing correction
All traits were tested for associations to cis- or trans-acting SNPs individually. Study-wise individual cutoff levels were calculated to test for cis- and trans-associations. To derive a study-wise cis-cutoff level at significance level α=0.05, we first determined all cis-acting SNPs within the data set. To allow for detailed comparisons of our results with those of Schadt et al.,16 we used the same definitions, that is, cis-acting SNPs are those that occur within 1 Mb upstream or downstream of the known or predicted 5′- and 3′-ends of any trait gene. Thus, the study-wise Bonferroni-adjusted P-value threshold equals 0.05/2 607 664=1.92 × 10−8, where 2 607 664 is the total number of possible cis-associations based on the above definition. Accordingly, the study-wise trans-specific Bonferroni’s cutoff level was computed as the total number of tests for trans-associations, that is, 0.05/4.6 billion=1.08 × 10−11.
Further analysis of SNPs, haplotypes and genetic linkage
Information on SNPs was obtained from the dbSNP database37 (http://preview.ncbi.nlm.nih.gov/snp). Polymorphisms occurring in an eQTL were tested for pairwise linkage to SNPs in the neighborhood by tagging specific blocks of genetic linkage using Haploview38 (V4.2, HapMap V3R2) or SNAP39 (http://www.broadinstitute.org/mpg/snap; using HapMap release 22 and 1000 genomes Pilot1) for CEU samples each. Pairwise r2 was used as measure for linkage disequilibrium (LD). The genomic regions of trait genes were screened for previously described copy number variations using the genome variation database from the Center of Applied Genomics (http://projects.tcag.ca/variation/).
Results
GWA analysis and extraction of ADME genes
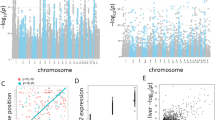
We used additive linear models in GenABEL34 to detect associations between SNPs and expression traits, and we considered a comprehensive set of available clinical data as covariates (Supplementary Table S1). In total, we identified 1226 significant associations (1179 cis and 47 trans) between 1163 SNPs and 371 different expression traits (Figure 2, Supplementary Table S2).

Venn diagram of eQTL results in the Stuttgart and Seattle studies. Comparison of significant genetic associations detected in the two studies after Bonferroni's correction. Numbers in the upper box refer to genome-wide associations (GWAs), whereas numbers in the lower box refer to ADME genes. The number of associated expression traits is given in brackets. ADME, absorption, distribution, metabolism and excretion; eQTL, expression quantitative trait loci.
As we were particularly interested in genes relevant to drug disposition and toxicity, we filtered identified eQTLs according to their potential relevance in the context of ADME processes. We compiled an ADME gene set comprising 682 genes (Supplementary Table S3) from various resources including the PharmaADME Working Group list of ADME genes (http://pharmaadme.org/), the NURSA (Nuclear Receptor Signaling Atlas) Consortium40 and the PharmGKB knowledge base41 (http://www.pharmgkb.org/). By requesting either SNP or trait gene or both to be an ADME gene, we identified a total of 95 significant associations, of which 89 were cis- or presumably cis-acting and 6 were trans- or presumably trans-acting. Table 1 shows 21 associations that could be validated by comparison with the Seattle study (see below), and Table 2 shows the other 74 ADME associations uniquely identified in our study.
Comparison and validation of eQTLs
To investigate reproducibility of our eQTL results, we performed a detailed comparison with the Seattle study,16 which differed in several important aspects including technology platform, origin and number of samples analyzed and available medical information. Surprisingly, only 200 of all 1262 associations (16%) were also found in the Seattle study when Bonferroni's adjustment was applied to both studies (Figure 2). A more detailed analysis showed that 30 of the 200 replicated associations were exact validations, that is, the same SNPs were found in both studies to be associated with the same trait, whereas 170 SNPs were validated by LD, that is, they were found to be tight linkage (r2>0.8) to SNPs of the Seattle study. The remainder of the 1062 SNPs represented independent associations only found in our study (Supplementary Table S2). Remarkably, none of the trans-associations in either study was replicated by the other (Figure 2). Reanalysis of the data after omitting most of the covariates except those considered in the former study revealed 1313 associations, whereas still only ∼20% of all associations were found in both studies (data not shown).
Investigation of ADME eQTLs
Table 1 summarizes 21 of the 95 ADME associations that represent replications, including 3 exactly replicated eQTLs and 18 matches by LD, together mapping to 11 different trait genes.
For example, two SNPs in a haplotype block at the CYP3A locus on chromosome 7 were strongly correlated with increased expression of CYP3A5 in heterozygous carriers (see box plot in Figure 3). One of them (rs10242455) represents an exact replication and the other (rs1859690) a replication by LD (Table 1). Both of them were completely linked to each other and to rs776746, the causative SNP of the well-investigated CYP3A5*3 splice variant.42, 43 Box plots showing the genotype–phenotype correlation for one representative SNP of each trait are compiled in Figure 3. Further analyses regarding these replicated results are presented in Supplementary Results and Discussion and in Supplementary Table S4.

Box plots of validated ADME associations. Box and whisker diagrams include smallest gene expression values, lower quartiles, medians, upper quartiles, largest gene expression values and outliers of the 11 validated ADME associations. The size of each genotyping group is given in brackets along the x axis. ADME, absorption, distribution, metabolism and excretion.
Table 2 shows the 74 ADME associations (68 cis-acting and 6 trans-acting) uniquely identified in our study, which mapped to 31 different trait genes. Closer analysis showed that the corresponding SNPs had indeed been analyzed but were not significantly associated with expression in the former study.16 These unique associations included additional SNPs for traits mentioned in Table 1 but with LD of r2<0.8 (for example, ARNT, several GSTs, VKORC1) and additional ADME and ADME-related genes including the histamine and diamine-oxidizing copper enzyme ABP1, the arachidonic acid-metabolizing CYP4F12, the flavin monooxygenase FMO4, thymodylate synthase (ENOSF1/TYMS) involved in 5-fluorouracil response and the solute carrier SLC22A10, among others. Furthermore, UGT1A1 expression was associated with rs2070959 (located in UGT1A6), which is closely linked (r2=0.87) to rs8175347, the causal SNP of the UGT1A1*28 allele.44 Remarkably, this well-known polymorphism had not been detected by the Seattle study.16 Box plots of these unique eQTLs represented by one SNP each are shown in Supplementary Figure S1.
The only true trans-association significantly identified among ADME genes comprised six SNPs in the plasminogen (PLG) gene on chromosome 6, a zymogen of the serine protease plasmin, which were associated to expression of a distant serine protease UNQ9391(PRSS55) on chromosome 8. As depicted in Figure 4, three of the variants (rs1406891, rs783145 and rs1247558) are simultaneously locally associated with expression of PLG itself, thus substantiating functional impact. Whereas rs1247558 and rs1406891 are located in intergenic regions, rs783145 is located in an intron, suggesting that this or a closely linked variant might be the causative SNP.

Manhattan plot of novel trans-eQTL and related box-plots of cis/trans-associations. (a) Negative log-transformed P-values of all SNPs tested for association to the serine protease UNQ9391 (PRSS55), which is located on chromosome 8. A set of 6 trans-acting SNPs at the plasminogen (PLG) locus on chromosome 6 was identified to be significantly associated. The dashed line represents the P-value cutoff level for trans-associations. The three indicated SNPs are simultaneously locally associated with the expression of PLG itself. (b) Box plots showing cis (left) and trans (right) genotype–phenotype relationships for one selected SNP. eQTL, expression quantitative trait loci; SNP, single-nucleotide polymorphism.
Discussion
Several studies11, 12, 13, 14, 15, 16, 17, 18, 19 have shown the utility of eQTL analysis to elucidate relationships between genetic polymorphisms and gene expression on a genome-wide scale, thus contributing to understanding the genetic basis of inter-individual variability and heritability of complex traits. However, limited information is so far available regarding completeness, reproducibility and interpretation of such data.
Our study was performed in the human liver with a focus on ADME genes, which have not been given special attention in previous studies. It replicates a former study by Schadt et al.16 (the Seattle study) in terms of general approach and tissue analyzed, and we therefore strived to compare the results of the two studies in detail.
The fraction of replicated observations between this and the former study was between 16 and 20%, which included replications by LD. As these represent eQTLs replicated in two different studies, they should be highly reliable, justifying in-depth analysis without further validation. However, the large discrepancy between the two studies was unexpected. Likely reasons include technical differences between the two studies, in particular the different gene expression profiling platforms applied in our (Illumina) and the former study (Agilent Technologies), which used different probes for most genes. This can lead to different expression data, given the common occurrence of variant transcripts, SNP-interference with probe hybridization and inconsistent gene annotation.45, 46 Additional technical differences concern the genotyping arrays and differences in data preprocessing.
Moreover, it should be noted that we applied an additive genetic model, which implies a gene-dose effect, but also reveals recessive or dominant associations at lower significance. Importantly, these three types of associations represent the biologically most meaningful possibilities. In contrast, the eQTL association analysis by Schadt et al.16 was based on Kruskal–Wallis tests, that is, a codominant model, which also reveals extreme deviations from additivity, that is, overdominant associations in which the two homozygous groups show similar expressions but the heterozygotes differ significantly. This (unknown) fraction of the eQTL associations reported by Schadt et al.16 must therefore be expected to be not reproducible by our study.
Additional important differences between the two studies relate to the number, origin and sampling of tissue, as well as medical information available. Sample-related differences should be of uttermost importance, although this is quite difficult to prove as no direct comparison was possible. The Stuttgart cohort consisted exclusively of liver tissue removed surgically from Caucasian donors in one hospital, using only one procedure for sample collection, freezing, storage, RNA isolation, quality assessment, DNA isolation and microarray analysis. Although all but one sample were resected because of liver cancer, this fact by itself should not affect genotype–phenotype relationships because only non-tumorous material was analyzed. In contrast the 427 samples of the Seattle cohort consisted mostly of postmortem material obtained from prospective organ donors who were presumably cancer-free, but the tissue quality may vary more widely because of warm ischemia before preservation and long storage times before cryopreservation. Furthermore, the Seattle cohort was collected in three independent centers, giving rise to differences among samples regarding tissue acquisition and storage protocols, criteria for RNA quality, etc. Another significant influential factor in the Seattle study was ethnicity,16 in contrast to our study in which no influence was detected using population stratification methods. Finally, the availability of medical information was also different in the two studies. Whereas the former study used imputation methods to complete large parts of missing information, medical documentation for the Stuttgart liver samples were almost complete and comprised a larger number of parameters.
Taken together, the many potentially influential parameters that differed between the two studies may well explain why only a small fraction of eQTL results were reproducible. Given the difficulty in validating cis-associations, it is probably not surprising that none of the trans-associations could be reproduced, as these are based on additional indirect downstream effects and are therefore generally less well replicated than cis-associations.16, 47
Pharmacologically relevant eQTLs were extracted based on a comprehensive compilation of 682 non-overlapping ADME genes, resulting in 89 cis- and 6 trans-associations. Similarly to the entire eQTL set, only 3 of the 95 ADME eQTLs were exactly replicated, and another 10 eQTLs were shown to be replications by LD. More detailed information on all replicated associations is presented in Supplementary Results and Discussion. Several of the unique ADME gene associations of our study concern known polymorphically expressed genes, including UGT1A1,44 CYP4F1248 and GST subforms M3, O2 and T1. Although these were not identified in the Seattle study, they likely relate to these biologically confirmed polymorphisms. Except for CYP3A5 and CYP4F12, no other CYP genes were present among the identified eQTLs. The failure to detect the well-known polymorphic CYPs (for example, 2A6, 2B6, 2C19, 2D6) may be explained by the mechanisms involved which often include complex splicing events which may not be detectable by the microarray probes.49
During revision of this manuscript, another paper appeared,50 which similar to our paper compared the results of an eQTL association analysis in human livers (two collectives with n1=206 and n2=60) with the Seattle study.16 Using different validation sets, they found replication rates between 49 and 58% for cis-eQTLs, that is, about half of all cis-associations failed to be validated in their study. A thorough investigation of the factors influencing reproducibility revealed similar reasons as those mentioned in our study. The lower replication rate in our study compared with50 may be explained by more relaxed validation criteria in their study, in particular regarding muss less stringent multiple correction conditions. Other likely reasons for differing replication rates include the factors discussed above, that is, origin and sampling conditions of the tissues, statistical modeling, as well as ethnicity and other covariates of the liver donors.
In conclusion, we carried out a GWA analysis of >300 000 SNPs and 48 000 expression phenotypes determined in a cohort of 149 human surgical liver samples obtained from Caucasian donors to identify genetic loci associated with quantitative changes in gene expression. A subset of 95 significant genotype–phenotype relationships, which was classified as ADME or ADME-related may be of particular relevance for drug disposition and toxicity. Detailed comparison between this study and the similar Seattle study demonstrated that quantitative trait loci are difficult to reproduce because of a number of technical and statistical reasons, and that several studies are required to discover the full extent of genetic determination in quantitative traits. Follow-up studies to elucidate the causal variants and their biological and pharmacological relevance should therefore concentrate on the validated results.
Accession codes
References
Gilad Y, Rifkin SA, Pritchard JK . Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet 2008; 24: 408–415.
Cheung VG, Spielman RS . Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat Rev Genet 2009; 10: 595–604.
Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 2009; 41: 25–34.
Meyer UA . Pharmacogenetics and adverse drug reactions. Lancet 2000; 356: 1667–1671.
Daly AK . Genome-wide association studies in pharmacogenomics. Nat Rev Genet 2010; 11: 241–246.
Motsinger-Reif AA, Jorgenson E, Relling MV, Kroetz DL, Weinshilboum R, Cox NJ et al. Genome-wide association studies in pharmacogenomics: successes and lessons. Pharmacogenet Genomics 2012; doi: 10.1097/FPC.0b013e32833d7b45 (in press).
Franke L, Jansen RC . eQTL analysis in humans. Methods Mol Biol 2009; 573: 311–328.
Rockman MV, Kruglyak L . Genetics of global gene expression. Nat Rev Genet 2006; 7: 862–872.
Mackay TF, Stone EA, Ayroles JF . The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 2009; 10: 565–577.
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009; 106: 9362–9367.
Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L et al. A survey of genetic human cortical gene expression. Nat Genet 2007; 39: 1494–1499.
Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KCC et al. A genome-wide association study of global gene expression. Nat Genet 2007; 39: 1202–1207.
Göring HHH, Curran JE, Johnson MP, Dyer TD, Charlesworth J, Cole SA et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat Genet 2007; 39: 1208–1216.
Veyrieras J-B, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 2008; 4: e1000214.
Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C et al. Population genomics of human gene expression. Nat Genet 2007; 39: 1217–1224.
Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol 2008; 6: e107.
Heinzen EL, Ge D, Cronin KD, Maia JM, Shianna KV, Gabriel WN et al. Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol 2008; 6: e1.
Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J et al. Genetics of gene expression and its effect on disease. Nature 2008; 452: 423–428.
Webster JA, Gibbs JR, Clarke J, Ray M, Zhang W, Holmans P et al. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet 2009; 84: 445–458.
Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS et al. Genetic analysis of genome-wide variation in human gene expression. Nature 2004; 430: 743–747.
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai S-L et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet 2010; 6: e1000952.
Nies AK, Koepsell H, Winter S, Burk O, Klein K, Kerb R et al. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 2009; 50: 1227–1240.
Hofmann MH, Blievernicht JK, Klein K, Saussele T, Schaeffeler E, Schwab M et al. Aberrant splicing caused by single nucleotide polymorphism c.516G>T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J Pharmacol Exp Ther 2008; 325: 284–292.
Simòn-Sànchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D et al. Genome-wide association study reveals genetic risk underlying Parkinsons disease. Nat Genet 2009; 41: 1308–1312.
Smyth G . Limma: Linear models for microarray data. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S (eds). Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer: New York, 2005, pp 397–420.
Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R et al. Missing value estimation methods for DNA microarrays. Bioinformatics 2001; 17: 520–525.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5: R80.
Li Y, Willer C, Sanna S, Abecasis G . Genotype imputation. Annu Rev Genomics Hum Genet 2009; 10: 387–406.
Fransen K, Visschedijk MC, van Sommeren S, Fu JY, Franke L, Festen EAM et al. Analysis of SNPs with an effect on gene expression identifies UBE2L3 and BCL3 as potential new risk genes for Crohn's disease. Hum Mol Genet 2010; 19: 3482–3488.
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, Marchand M et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet 2009; 41: 89–94.
Tian C, Gregersen PK, Seldin MF . Accounting for ancestry: population substructure and genome-wide association studies. Hum Mol Genet 2008; 17 (R2): R143–R150.
Karacaoren B, Silander T, Alvarez-Castro J, Haley C, de Koning DJ . Association analyses of the MAS-QTL data set using grammar, principal components and Bayesian network methodologies. BMC Proceedings 2011; 5 (Suppl 3): S8.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D . Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM . GenABEL: an R library for genome-wide association analysis. Bioinformatics 2007; 23: 1294–1296.
Shinka T, Naroda T, Tamura T, Sasahara K, Nakahori Y . A rapid and simple method for sex identification by heteroduplex analysis, using denaturing high-performance liquid chromatography (DHPLC). J Hum Genet 2001; 46: 263–266.
McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008; 9: 356–369.
Smigielski EM, Sirotkin K, Ward M, Sherry ST . dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res 2000; 28: 352–355.
Barrett JC . Haploview: visualization and analysis of SNP genotype data. Cold Spring Harb Protoc 2009; 2009, pdb ip71.
Johnson AD, Zhang Y, Papp AC, Pinsonneault JK, Lim J-E, Saffen D et al. Polymorphisms affecting gene transcription and mRNA processing in pharmacogenetic candidate genes: detection through allelic expression imbalance in human target tissues. Pharmacogenet Genomics 2008; 18: 781–791.
Lanz RB, Jericevic Z, Zuercher WJ, Watkins C, Steffen DL, Margolis R et al. Nuclear receptor signaling atlas ( www.nursa.org ): hyperlinking the nuclear receptor signaling community. Nucleic Acids Res 2006; 34 (Database issue): D221–D226.
Altman RB . PharmGKB: a logical home for knowledge relating genotype to drug response phenotype. Nat Genet 2007; 39: 426.
Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet 2001; 27: 383–391.
Hustert E, Haberl M, Burk O, Wolbold R, He YQ, Klein K et al. The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics 2001; 11: 773–779.
Bosma PJ, van der Meer IM, Bakker CT, Hofman A, Paul-Abrahamse M, Witteman JC . UGT1A1*28 allele and coronary heart disease: the Rotterdam Study. Clin Chem 2003; 49: 1180–1181.
Barbosa-Morais NL, Dunning MJ, Samarajiwa SA, Darot JFJ, Ritchie ME, Lynch AG et al. A re-annotation pipeline for Illumina BeadArrays: improving the interpretation of gene expression data. Nucleic Acids Res 2010; 38: e17.
Dunning MJ, Curtis C, Barbosa-Morais NL, Caldas C, Tavare S, Lynch AG . The importance of platform annotation in interpreting microarray data. Lancet Oncol 2010; 11: 717.
Michaelson JJ, Loguercio S, Beyer A . Detection and interpretation of expression quantitative trait loci (eQTL). Methods 2009; 48: 265–276.
Cauffiez C, Klinzig F, Rat E, Tournel G, Allorge D, Chevalier D et al. Human CYP4F12 genetic polymorphism: identification and functional characterization of seven variant allozymes. Biochem Pharmacol 2004; 68: 2417–2425.
Zanger UM, Turpeinen M, Klein K, Schwab M . Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem 2008; 392: 1093–1108.
Innocenti F, Cooper GM, Stanaway IB, Gamazon ER, Smith JD, Mirkov S et al. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet 2011; 7: e1002078.
Acknowledgements
This study was supported by the German Federal Ministry of Education and Research (BMBF Virtual Liver Network grants 0315755 and 0315756) and the Robert-Bosch Foundation, Stuttgart, Germany.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Schröder, A., Klein, K., Winter, S. et al. Genomics of ADME gene expression: mapping expression quantitative trait loci relevant for absorption, distribution, metabolism and excretion of drugs in human liver. Pharmacogenomics J 13, 12–20 (2013). https://doi.org/10.1038/tpj.2011.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2011.44
Keywords
This article is cited by
-
From molecular mechanisms of prostate cancer to translational applications: based on multi-omics fusion analysis and intelligent medicine
Health Information Science and Systems (2023)
-
Epistatic interactions of genetic loci associated with age-related macular degeneration
Scientific Reports (2021)
-
Comprehensive Transcriptome Analyses in Sea Louse Reveal Novel Delousing Drug Responses Through MicroRNA regulation
Marine Biotechnology (2021)
-
The drug likeness analysis of anti-inflammatory clerodane diterpenoids
Chinese Medicine (2020)
-
Genome-wide pQTL analysis of protein expression regulatory networks in the human liver
BMC Biology (2020)
