
Abstract
Prenatal infection is an environmental risk factor for various brain disorders with neurodevelopmental components, including autism spectrum disorder and schizophrenia. Modeling this association in animals shows that maternal immune activation negatively affects fetal brain development and leads to the emergence of behavioral disturbances later in life. Recent discoveries in these preclinical models suggest that epigenetic modifications may be a critical molecular mechanism by which prenatal immune activation can mediate changes in brain development and functions, even across generations. This review discusses the potential epigenetic mechanisms underlying the effects of prenatal infections, thereby highlighting how infection-mediated epigenetic reprogramming may contribute to the transgenerational transmission of pathological traits. The identification of epigenetic and transgenerational mechanisms in infection-mediated neurodevelopmental disorders appears relevant to brain disorders independently of existing diagnostic classifications and may help identifying complex patterns of transgenerational disease transmission beyond genetic inheritance. The consideration of ancestral infectious histories may be of great clinical interest and may be pivotal for developing new preventive treatment strategies against infection-mediated neurodevelopmental disorders.
Similar content being viewed by others
Introduction
Disruption of normal brain development is implicated in a number of neuropsychiatric diseases with neurodevelopmental origins, including schizophrenia, autism spectrum disorder and bipolar disorder.1 One commonality between these disorders is that the underlying pathological processes likely start early during fetal development and continues during subsequent brain maturation.2 The disruption of neurodevelopmental and maturational processes can lead to widespread and long-lasting abnormalities in central nervous system structure and functions. Typically, neurodevelopmental disorders are characterized by a heterogeneous group of symptoms affecting cognitive functions, emotions and behavior, thereby undermining basic human processes of perception and judgment. In view of their chronicity and severity, neurodevelopmental disorders impose tremendous burdens on the affected individuals, and in turn, on their families and society in general.3
Besides genetic contributions,4 various environmental factors appear to increase the risk for neurodevelopmental disorders.1, 5 Many of these factors operate during critical periods of central nervous system development. One of these factors is prenatal exposure to infection, which is increasingly recognized to play an important etiological role in various brain disorders with neurodevelopmental components, including schizophrenia,5, 6 autism spectrum disorder7, 8 and bipolar disorder.6, 9 Hence, prenatal exposure to immune challenges may be best viewed as a general vulnerability factor for neurodevelopmental brain disorders rather than a disease-specific risk factor.10, 11 Indeed, the adverse effects induced by prenatal infection may reflect an early entry into a deviant neurodevelopmental route, but the specificity of subsequent disease or symptoms is likely to be influenced by the genetic and environmental context in which the prenatal infectious process occurs. This notion is consistent with the emerging evidence suggesting that seemingly remote disorders, such as schizophrenia, autism spectrum disorder and attention-deficit/hyperactivity disorder share considerable amounts of risk factors and brain dysfunctions.10, 11 The presence of shared genetic and environmental risks among these illnesses has led to the proposal that they might lie along a continuum of genetically and environmentally induced neurodevelopmental causalities,1, 12 wherein prenatal infection may be one of the many factors that shape the eventual pathological outcomes.
The epidemiological link between prenatal infection and increased risk of neurodevelopmental brain disorders also receives strong support from experimental work in animal models.13 On the basis of human epidemiological findings, a number of translational rodent models have been established to explore the consequences of prenatal immune activation on brain and behavioral development. These models are based on maternal gestational exposure to specific infectious agents such as influenza virus or immune activating agents such as the bacterial endotoxin lipopolysaccharide or the viral mimic poly(I:C) (=polyriboinosinic–polyribocytidylic acid). Converging evidence from these models suggests that prenatal immune activation can negatively affect early fetal brain development and change the offspring’s neurodevelopmental trajectories, which in turn can lead to the emergence of behavioral and cognitive disturbances in later life.13, 14, 15, 16 The long-term behavioral and cognitive abnormalities induced by prenatal immune challenges in rodent models have already been extensively reviewed elsewhere (for review see Meyer,13 Meyer and Feldon,15 and Estes and McAllister17) and have recently been extended to models using primates with more advanced cortical development and functions.18, 19, 20
Despite the advances in this research field, the mechanisms whereby prenatal infection leads to long-term effects on central nervous system functions are only partially understood. In keeping with the crucial role of epigenetic remodeling in early development, it has recently been postulated that epigenetic modifications may be a critical molecular mechanism by which prenatal immune activation can mediate changes in brain development and functions.21, 22 The present review summarizes and integrates novel findings supporting this hypothesis and highlights how infection-mediated epigenetic reprogramming could be a molecular mechanism shaping disease risk across generations.
Pathophysiological processes induced by maternal infection
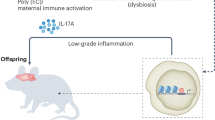
Infection during pregnancy induces a spectrum of pathophysiological changes in the maternal host and fetal compartments.23 Figure 1 provides a schematic summary of the most relevant pathophysiological processes associated with maternal infection during pregnancy. The nature of these processes, and the resulting neurodevelopmental sequelae in the offspring, are likely influenced by a number of factors, including virulence of the pathogen, timing of infection and (genetic) susceptibility of the host. Interestingly, increased risk for neurodevelopmental disorders is observed following prenatal exposure to a variety of pathogens, including influenza, rubella, measles, polio and others (for review see Brown and Derkits,5 and Silasi et al.23). Only some of these pathogens are vertically transmitted and directly invade the fetal system. One prominent example showing transplacental transmission is rubella, which has long been known to be responsible for a spectrum of developmental abnormalities referred to as the congenital rubella syndrome.24 The majority of pathogens, however, stay refined to the maternal host upon infection during pregnancy.23 Even when lacking vertical transmission, maternal infection during pregnancy can induce long-term effects on the offspring, suggesting that the association between prenatal infection and neurodevelopmental disorders is, to a large extent, driven by factors that are common to all infections. In support of this notion, rodent studies provide substantial evidence for the hypothesis that inflammatory cytokines are one of the key players in re-wiring ongoing developmental processes into pathological trajectories following prenatal infection.25, 26, 27 Cytokines are increased during the acute phase response to infection and are important mediators of the host’s defense against infectious agents.28, 29 In the context of prenatal infection, some of the maternally produced cytokines can be transferred via the placenta, as shown in animals and human placental tissue,30, 31 and contribute to cytokine imbalances in the fetal system. In addition, the placenta and fetal organs can mount increased cytokine production in response to maternal infection.32, 33, 34

Post-acute pathophysiological effects of maternal infection during pregnancy. Some infectious pathogens (for example, rubella virus, cytomegalovirus, herpes simplex virus-2 and Toxoplasma gondii) can be vertically transmitted and lead to congenital infections, which in turn can result in severe developmental deficits. Maternal infection during pregnancy can further induce a number of pathophysiological responses, even if the pathogen is not vertically transmitted. These responses include the production of soluble immune factors such as cytokines and other mediators of inflammation, as well as reactive oxygen species. Some of these factors might cross the placental barrier and enter the fetal environment, thereby causing fetal inflammation and oxidative stress. Maternal infection during pregnancy can further induce inflammatory response in the placenta and cause placental insufficiency, which in turn can cause fetal hypoxemia. In addition, infection can cause (temporary) states of macronutrient and micronutrient deficiency, which limits the fetal availability of essential nutrients necessary for normal fetal development and growth. Finally, maternal infection during pregnancy can modify the microbial composition of the placenta, which might alter the development of the offspring’s microbiome (not shown).
The post-acute effects of maternal infections are not limited to cytokine-associated inflammatory responses but further involve other pathophysiological processes, including nutrient deficiency and oxidative stress (Figure 1). Oxidative stress refers to an increase in production and/or decrease in elimination of reactive oxygen species. Reactive oxygen species are released as part of an activation cascade of innate immune cells to kill invading pathogens35 and can exert cytotoxic effects. Similar to cytokines, reactive oxygen species levels typically rise in response to an infection, which in turn can trigger oxidative stress in the fetal environment and negatively affect fetal brain development, both in humans and in animal models.36
Reduced availability of macro- and micronutrients for the fetus is yet another important post-acute effect of maternal infection (Figure 1). The infected host usually displays signs of sickness behavior, which is characterized by fever, malaise and reduced water and food intake.37 Reduction in food intake might indeed be of great relevance in the context of prenatal infection, with several studies highlighting a significant link between prenatal malnutrition and pathological brain development.38, 39 Studies in rats and mice suggest that the association between prenatal malnutrition and neurodevelopmental disorders might result, at least in part, from a reduction in the availability of micronutrients such as zinc and iron, which are crucial for normal brain development.40, 41
Finally, it should be noted that infection-induced pathophysiological changes in the placenta might amplify the response to maternal infection and the downstream pathological effects on the fetus. The placenta plays a key role in maternal–fetal interactions and expresses a variety of factors important for the homeostatic response to infectious agents.42 Maternal infections also typically disrupt the circulation and architecture of the placenta,27, 43, 44 as shown by studies in rodents and humans, which in turn can induce placental insufficiency and reduce the delivery of oxygen and essential nutrients to the developing offspring (Figure 1).
In summary, infection during pregnancy induces a number of post-acute pathophysiological processes in the maternal host, which in turn can all contribute to the disruption of normal brain development. As discussed in detail below, novel findings suggest that epigenetic modifications may represent an important molecular mechanism whereby these post-acute pathophysiological processes are translated into long-term brain abnormalities, even across generations.
A novel role of epigenetic modifications in infection-mediated neurodevelopmental disorders
A short overview of epigenetic mechanisms
‘Epigenetics’ refers to the combination of mechanisms that confer long-term and heritable changes in gene expression without altering the DNA sequence itself.21 Epigenetic programming is dynamic and responsive to different environmental exposures during development and includes several interrelated processes (Figure 2), including chromatin remodeling, histone modifications, DNA methylation and expression of microRNAs (miRNAs).21, 22, 45 Histone modifications are covalent post-translational modifications of histone proteins, which include, among others, methylation, phosphorylation, acetylation, ubiquitylation and sumoylation.21, 22, 45 Histone modifications can define the extent to which DNA is wrapped around the nucleosome core, thereby influencing the accessibility of the gene transcription machinery and subsequent gene expression. DNA methylation, on the other hand, consists of covalent methylation of cytosine rings that are found at cytosine–phosphodiester–guanine (CpG) dinucleotides.21, 22, 45 When located in distinct genomic regions such as gene promoter or enhancer sites, DNA methylation typically acts to repress gene transcription.21, 22, 45 Silencing of gene transcription by DNA methylation can be mediated by direct interference with the binding of transcription factors or enhancers to recognition elements that contain CpG dinucleotides, or through recruitment of methylated DNA-binding factors that in turn attract chromatin-inactivating complexes including histone deacetylases and histone methyltransferases. Finally, miRNAs are a class of small non-encoding RNAs (~22 nucleotides long) that can control target gene expression post-transcriptionally.21, 22, 45 Interestingly, recent studies have demonstrated that epigenetic mechanisms, such as DNA methylation, not only regulate the transcription of protein-encoding genes, but also the expression of miRNAs.46 Conversely, miRNAs can control the expression of important epigenetic regulators, including DNA methyltransferases and histone deacetylases. Hence, there is a dynamic regulatory network between different epigenetic pathways, which altogether organize gene expression profiles through transcriptional or post-transcriptional mechanisms.21, 22, 45, 46

Schematic representation of major epigenetic mechanisms. The DNA–protein complex is referred to as chromatin. The functional unit of the chromatin is the nucleosome, which is composed of DNA wrapped around a core octamer of histone proteins. The DNA–histone interaction occurs at the N-terminal tails of these histones, which face outward and are sites for epigenetic marking known as histone modifications. These modifications represent a first major epigenetic mechanism modulating gene expression and involve methylation, phosphorylation, acetylation, ubiquitylation and sumoylation. DNA methylations at cytosine rings, typically found at CpG dinucleotides, are another important epigenetic mark that can influence gene expression. Finally, micro-RNAs (miRNAs), a class of small, non-coding RNAs, can control target gene expression post-transcriptionally. CpG, cytosine–phosphodiester–guanine.
How can epigenetic modifications influence brain development?
A growing number of studies highlight the importance of epigenetic mechanisms in normal brain development.45 Interestingly, transcriptional changes in the brain occur more frequently during prenatal life as compared to any other age in life.47 A temporally precise and specific regulation of gene expression thus seems indispensable for normal brain development. This regulatory process involves methylation-related epigenetic modifications, which allow a fine-tuning of fetal gene expression according to specific stages of brain development.48, 49, 50 A critical role for DNA methylation in early brain development is also supported by the dynamic expression of DNA methyltransferases during prenatal life,51 and by studies using transgenic mouse models showing neurodevelopmental deficits in mice with mutations in the methyl-CpG-binding protein 2 (Mecp2) gene.52, 53 Furthermore, mice lacking distinct DNA methyltransferases (DNMTs) in the forebrain have been found to display severe learning and memory deficits, as well as impaired synaptic plasticity.54, 55
In addition to epigenetic processes involving DNA methylation, several other epigenetic mechanisms appear to be critical for neuronal development and functions. For example, it has been shown that histone modifications can regulate the conversion of oligodendrocytes into neural stem cells.56 Furthermore, Fischer et al.57 demonstrated in a mouse model of neurodegenerative disorders that increasing histone acetylation could promote synaptogenesis and augment cognitive functions.
Accumulating evidence suggests that miRNAs may similarly play a role in central nervous system development. For example, it has been shown that miRNAs in specific neuronal or glial cell populations show a dynamic expression pattern during brain development.58, 59 Moreover, a recent study reported distinct temporal patterns of miRNA expression in the brain throughout gestation, and from early neonatal to adult life.60 The developmentally regulated pattern of miRNA expression is indicative of a functional role of these molecules in normal brain development. However, our understanding of how miRNA can influence neurodevelopmental processes is still in its infancy and warrants further investigations.
The importance of epigenetic mechanisms in brain development is also supported by a plethora of findings demonstrating epigenetic alterations in neurodevelopmental disease models that are based on exposure to environmental adversities in early life. As reviewed in detail elsewhere21, 45, 61 stable epigenetic modifications may represent an important mechanism by which exposures to early-life environmental adversities can induce pathological consequences, even across multiple generations. Transgenerational transmission of disease susceptibility or epigenetic modification has been observed following early-life exposure to various environmental adversities (see Table 1 for prenatal environmental adversities), including prenatal or neonatal stress,62, 63, 64, 65, 66, 67, 68, 69 prenatal malnutrition,70, 71, 72, 73, 74, 75 endocrine disruptors76, 77, 78, 79, 80, 81 and chronic psychostimulant or alcohol intake.82, 83 The phenomenon of non-genetic transgenerational transmission of behavioral traits has gained increasing recognition in view of its potential importance in the etiology and treatment of multifactorial disorders.61, 84, 85 As discussed in the subsequent sections, recent research now suggests that similar effects can be induced by prenatal exposure to infection.
Epigenetic and transgenerational effects of prenatal infection
Epigenetic modifications induced by prenatal infection
Several recent studies sought to examine the putative effects of prenatal infection on epigenetic processes. Using a rat model of poly(I:C)-induced maternal immune activation, Hollins et al.86 revealed that prenatally infected offspring exhibited significant differences in the expression of miRNA in the entorhinal cortex, a brain area implicated in neurodevelopmental disorders. Interestingly, a large subset of these miRNAs were clustered within the Dlk1-Dio3-imprinted domain on 6q32, which is associated with schizophrenia, and were predicted to regulate pathways involved in synaptic remodeling, learning and memory formation.86 Maternal immune activation by poly(I:C) treatment in mice has also been found to alter histone modifications in the offspring. More specifically, offspring of poly(I:C)-treated mice displayed changes in promoter-specific histone acetylation and corresponding transcriptional changes, the latter of which affected genes associated with neuronal development, synaptic transmission and immune signaling.87
Accumulating evidence suggest that prenatal infection can also cause stable changes in DNA methylation. For example, Basil et al.88 found that prenatal exposure to the viral mimic poly(I:C) caused global changes in the level of DNA methylation in the adolescent mouse brain, including Mecp2 promoter hypomethylation. A recent study by Labouesse et al.89 extended those findings by assessing correlations between DNA-related epigenetic modifications, expression levels of corresponding genes and behavioral deficits. The authors showed that prenatal viral-like immune activation by poly(I:C) in mice induced methylation-related promoter remodeling of GAD1 and GAD2 in the prefrontal cortex. GAD1 and GAD2 encode for two isoforms of the rate-limiting enzyme for γ-aminobutyric acid biosynthesis. It was shown that offspring born to immune-challenged mothers displayed GAD1 and GAD2 promoter hypermethylation and associated reductions in the expression of the corresponding mRNA transcripts (GAD67 and GAD65), which in turn correlated with deficits in social interaction and impairments in working memory.89 These findings suggest that methylation-related epigenetic modifications at presynaptic GABAergic systems may represent a mechanism whereby maternal infection during pregnancy can induce long-term behavioral impairments in the offspring.
Using the same mouse model of poly(I:C)-induced maternal immune activation, a recent study by Richetto et al.90 examined genome-wide DNA methylation differences at single-nucleotide resolution by capture array bisulfite sequencing in the adult prefrontal cortex. It was shown that offspring of immune-challenged mothers displayed hyper- and hypomethylated CpGs at numerous loci and at distinct genomic regions.90 The differences in methylation were again associated with transcriptional changes of the corresponding genes, suggesting that the infection-induced epigenetic modifications had a functional impact on gene expression.90
Taken together, these findings indicate that prenatal infection can cause lasting changes in the offspring’s epigenome. The available data thus far show that viral-like maternal immune activation in early/middle (between gestation day (GD) 9 and 12)87, 88 or in late (GD15 and beyond)86, 89 gestation causes such changes. Recent evidence suggests, however, that the timing of viral-like immune challenge critically determines the specificity of infection-mediated epigenetic modifications,90 since early and late gestational window clearly differ in terms of methylation-related epigenetic modifications they induce.
Transgenerational effects of prenatal infection
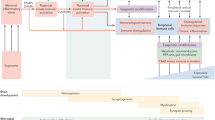
Recent studies extended the abovementioned findings by assessing whether behavioral and cognitive abnormalities emerging in the direct descendants (F1) of gestationally immune-challenged mothers could be transmitted across subsequent generations (F2 and F3) without any further immune exposures. Using the prenatal poly(I:C) administration model in mice, it was repeatedly shown that some of the behavioral abnormalities are not only present in the direct descendants of immune-challenged mothers, but are transmitted to the subsequent generations,91, 92 at least when the immune challenge was induced in early/middle pregnancy (that is, between GD9 and GD12). Interestingly, one study reported a transgenerational transmission of behavioral abnormalities mostly via the paternal lineage, which extended to the third (F3) generation of offspring (Figure 3).92 The paternal mode of transmission is consistent with other models of early-life adversities, such as pre- and neonatal stress and prenatal malnutrition.62, 63, 64, 65, 66, 70, 71 The fact that prenatal immune activation can cause transgenerational transmission of pathological phenotypes via the paternal lineage strongly suggests the involvement of epigenetic modifications in male gametes, which in turn could mediate epigenetic inheritance across generations.21, 45, 63, 66 Prenatal immune activation thus likely alters epigenetic marks in the germ line of the direct offspring, which resists erasure and epigenetic reestablishment during germ cell development.21

Summary of the transgenerational transmission and modification of behavioral deficits induced by prenatal immune activation. The use of a mouse model of viral-like immune activation, which was induced by maternal treatment with the viral mimetic poly(I:C), led to the recent discovery of transgenerational effects following prenatal immune activation (for details, see Weber-Stadlbauer et al.92). In this model, reduced sociability and increased fear-related behavior were similarly present in first-generation (F1) and second-generation (F2) offspring of immune-challenged ancestors. Sensorimotor gating impairments were confined to the direct descendants of infected mothers, whereas increased behavioral despair emerged as a novel phenotype in the second generation. The transgenerational effects were transmitted via the paternal lineage (not shown) and were stable until the third generation (F3), demonstrating transgenerational non-genetic inheritance of pathological traits following prenatal immune activation.
It remains to be determined further, however, why early-life adversities such as maternal infection largely spare the female germ cells. One possible explanation may relate to the differential developmental dynamics of male and female gametes. Germ cells start developing shortly after fertilization and rapidly proliferate until they migrate to the genital ridge, where sex determination occurs.21 Once the primordial germ cells are sexually differentiated, oocytes and male germ cells have different developmental dynamics; following sexual differentiation, the oocyte enters into meiosis and arrests until puberty, while male germ cells go into arrest until birth, when they undergo a phase of proliferation and then complete meiosis during puberty.93
In addition to its transgenerational effects on behavior, prenatal exposure to viral-like immune activation in mice was also found to modify transcriptional activity across generations.92 Using next-generation mRNA sequencing, it was shown that prenatal poly(I:C)-induced immune activation caused widespread gene expression changes in the brains of both the F1 and F2 generations. Intriguingly, while some transcriptional changes were uniquely present in either F1 or F2 offspring, others were common to both generations. Hence, the prenatal infection-induced transgenerational transmission of behavioral abnormalities (Figure 3) is associated with, and perhaps even mediated by, transgenerational modifications of gene expression.
Depending on the timing of the environmental insults or genetic loci, some of the epigenetic marks may affect only the germ line while sparing somatic tissues in the first generation. This may explain why certain behavioral deficits emerge as a novel phenotype only in the second and third generation of infected ancestors (Figure 3). Future studies are warranted in order to compare infection-induced epigenetic modifications in somatic cells and gametes. Such studies will help to further elucidate the mechanisms underlying the transgenerational modification and inheritance of brain pathology following prenatal immune activation.
Potential mechanisms through which prenatal infection might induce epigenetic changes (across generations)
The potential role of cytokines
As mentioned above, epidemiologic studies suggest that the risk of neurodevelopmental disorders is increased after prenatal exposure to a variety of viral, bacterial or protozoan pathogens, including those that are not vertically transmitted to the fetus. Based on these findings, it has been proposed that cytokine-associated inflammatory processes may be key in mediating the negative effects of maternal infection.94 Molecular work in mouse models of maternal immune activation has provided additional support for this hypothesis. For example, prenatal administration with specific cytokines such as interleukin-6 (IL-6) or IL-17a can mimic the long-term consequences of prenatal viral-like immune activation in mice.25, 26 Conversely, elimination of these cytokines from the maternal immune response by genetic interventions or blocking antibodies attenuates the long-term effects of prenatal immune challenge on brain and behavior.25, 26
Given these findings, it appears plausible that enhanced expression of inflammatory cytokines may be one of the factors that can directly alter epigenetic programs in the event of prenatal infection. In support of this notion, a study by Hodge et al.95 demonstrates that IL-6 increases the expression and biological activity of DNMT1. Some of the changes in DNMT1 activity may result from phosphorylation of the enzyme following exposure to IL-6, and the subsequent activation of downstream signaling pathways such as the AKT pathway.95 While most of the initial findings linking cytokines to epigenetic effects stem from experiments with cancer cell lines, it is likely that IL-6 regulates DNMT1 in several distinct cell types, including neurons and other brain cells. It was shown that IL-6 and DNMT1 can also influence cell migration,96 a process which is also essential in neurodevelopment. Recent studies also identified a direct influence of IL-17a on the epigenetic machinery through the modulation of histone deacetylases (HDACs). More specifically, IL-17a was shown to inhibit HDAC activity via the phosphoinositide-3-kinase (PI3K) pathway.97 Finally, one study found that TNF-alpha increases histone acetylation activity,98 suggesting that this inflammatory cytokine can act on the epigenetic machinery as well.
Exposure to prenatal inflammation stimuli has also been shown to cause Mecp2 promoter hypomethylation.88 Mecp2 is important for epigenetic regulation due to its ability to bind to methylated DNA and to chromatin modifying complexes such as HDACs. Therefore, Mecp2 is considered both an epigenetic ‘reader’ and a ‘writer’ (for review see Ausio99). Additional research is required, however, to examine more directly whether the effects of prenatal immune activation on Mecp2 promoter hypomethylation involves specific cytokines or other pathophysiological processes associated with inflammation.
Taken together, there is initial evidence supporting a role of cytokines in modulating the epigenetic machinery in the event of prenatal infection. Hence, increased fetal expression of inflammatory cytokines may be one of the upstream factors causing epigenetic reprogramming and associated neurodevelopmental changes in offspring of gestationally infected mothers.
The potential role of folic acid
As previously discussed, maternal infection typically leads to temporary reduced food consumption, thereby reducing nutrient availability for the developing organism (Figure 1). Besides its effects on other micronutrients such as zinc and iron, reduced intake of micronutrients may lead to a transient deficiency of folic acid. Folic acid was shown to play a key role in the modulation of inflammatory responses: supplementation of patients with folic acid inhibits pro-inflammatory responses, such as inhibiting chemokine secretion and reducing the elevated levels of reactive oxygen species.100 Vice versa, mouse monocytes, under conditions of folate deficiency, show a 2- to 3-fold increase in the expression of several inflammatory cytokines, such as IL1b, IL-6 and TNFα.101 Folic acid has also long been implicated in neurodevelopmental processes, as exemplified by the presence of spina bifida in offspring of mothers who were deprived from folic acid during pregnancy.102 Moreover, imbalances in folate levels during pregnancy represent an important, epidemiologically valid risk factor for later psychiatric disease.103 Besides its established etiological role in neurodevelopmental disorders,104 low levels of folate are also present in patients diagnosed with schizophrenia.105 There is a growing body of evidence linking folic acid deficiency with autism as well.106, 107, 108 Interestingly, maternal folic acid supplementation has been associated with beneficial effects on neurodevelopment and autism.103 Moreover, mothers of children with autism have a significantly lower mean folic acid intake during early pregnancy compared to mothers of children with autism spectrum disorder.107
Representing one of the major dietary sources for methyl groups,109 folic acid is likely to have a direct influence on epigenetic processes during normal prenatal development. Indeed, a recent study identified a striking correlation between maternal folic acid and methylation status of a variety of genes in the offspring, some of which are related to neurological functions and to embryonic development.110 In the pathological context of maternal infection, decreased availability of folic acid may affect methylation-related epigenetic processes in the fetus, thereby contributing to infection-mediated epigenetic programming. Even though biologically plausible, however, this hypothesis still awaits direct examination in future studies.
The potential role of the microbiome
The recent years have witnessed an increasing interest in the role of the microbiome in health and disease. The human body is composed of a complex ecosystem of microbial cells, approximating the same number as the number of human cells in the human body.111 There is a clear symbiotic link between host and microbe, wherein the microbiota is also key mediating for gene–environment interactions relevant to various physiological processes.112 Whereas initial studies mainly focused on obesity-related changes in the microbiome and its contribution to the disease phenotype, it has recently been shown that the microbiome can also influence behavior and contribute to brain development and function (for review see Cryan and Dinan113). While the fetal environment was initially thought to be entirely sterile, recent evidence suggests that some bacteria are present in the amniotic fluid114 and the placenta.115 One implication of these new discoveries is that the microbial composition of the developing offspring may be sensitive to environmental changes even during prenatal stages of life. In support of this notion, a recent study in mice demonstrated that prenatal viral-like immune activation alters the offspring’s composition of the gut microbiota.116 Notably, some of the main metabolic products of the gut microbiota are short-chain fatty acids, including sodium butyrate. Sodium butyrate can readily cross the blood–brain barrier and has been shown to modulate the expression of various genes in the prefrontal cortex, particularly those involved in neuronal excitation or inhibition.117 At the same time, sodium butyrate can interact with the epigenetic machinery by inhibiting HDACs.118 Therefore, aside from being a bacterial metabolite and a major source of energy for intestinal epithelial cells, sodium butyrate represents an efficient epigenetic regulator. Future studies are warranted in order to investigate whether infection-induced changes in gut microbiota may induce downstream changes in sodium butyrate, which in turn may affect the epigenetic machinery. Such changes would possibly also affect the fetus’ germ cells, and thereby, they could also contribute to the transgenerational transmission of behavioral traits induced by maternal immune activation.
The influence of the precise timing of maternal infection
An important aspect that warrants further investigation relates to the potential influence of the precise timing of prenatal immune activation, both in terms of its effects on the offspring’s epigenome and on the transgenerational transmission of pathological traits. Exposure to environmental insults during distinct gestational stages likely affects different neurodevelopmental events, developing brain regions, and cellular maturational processes, which in turn might shape the specificity of long-term brain dysfunctions in the offspring.34 In support of this notion, numerous rodent studies highlighted a critical influence of the precise timing of prenatal infection with regards to its long-term brain and behavior consequences in the offspring.119, 120
It has long been recognized that epigenetic processes follow timed developmental patterns during embryogenesis and fetal development.121, 122 It thus follows that maternal immune activation induces differential epigenetic modifications in the offspring depending on the precise timing of exposure. Indeed, a recent mouse study provided the first line of evidence suggesting that the epigenetic effects of prenatal immune activation are critically influenced by the timing of prenatal immune challenge.90 While immune activation in early and late gestation affected a relatively small number of common genes, the two prenatal conditions mostly showed methylation differences in distinct loci.90 Moreover, the differential epigenetic effects of early and late prenatal immune activation were associated with distinct transcriptional changes and behavioral abnormalities.90 Against this background, it appears essential to further explore whether and how timing-dependent effects are also reflected by the nature of the transgenerational effects induced by prenatal infection.
Conclusions
The investigation of possible immune mechanisms in neurodevelopmental disorders is a long-standing area of research that continuously attracts the attention from basic scientists and clinicians alike. Within this neuroimmune framework of brain disease, a great deal of interest has been centered on the possible contribution of infections in prenatal life. The recent outbreak of Zika virus and its accompanying risk for severe brain anomalies such as microcephaly123 has moved the risks of prenatal infection in the spotlight of public attention.
Modeling the consequences of prenatal infection in animals has led to the new discoveries that this early-life adversity induces stable epigenetic modifications in the offspring. There is increasing recognition of and evidence for altered epigenetic programming in the pathogenesis of neurodevelopmental disorders, including schizophrenia, autism and bipolar disorder.124, 125, 126, 127 Against these backgrounds, the novel findings of infection-mediated epigenetic modifications raise the intriguing possibility that prenatal exposure to immune challenges may be one of the environmental factors causing epigenetic abnormalities in these disabling brain disorders. Since at least some of these epigenetic modifications may be modifiable throughout life,22, 128 these findings may help to open new avenues for symptomatic or even preventive treatments in subjects with prenatal infectious histories.
The transgenerational transmission and modification of brain pathology following prenatal immune activation highlights a novel pathological aspect of this early-life adversity in shaping disease risk across generations. Given that prenatal infection is implicated in a variety of neurodevelopmental brain disorders, these new findings appear relevant to brain disorders independently of existing diagnostic classifications and may help identifying complex patterns of transgenerational disease transmission beyond genetic inheritance. These discoveries may further emphasize the importance for global health strategies in focusing on the development and promoting vaccinations, in particular for women who are pregnant or those who plan to become pregnant. Indeed, vaccinations may have the potential to prevent a wide spectrum of neuropsychiatric and neurological disorders associated with prenatal infection, and they may even prevent possible pathological effects across generations. Unfortunately, the limited access to vaccines in certain countries, along with the continuous trend against vaccinations within certain groups of the society, have led to a resurgence of serious infectious diseases, including measles, rubella and polio.129 Hence, there are obvious challenges for future attempts to implement vaccinations as preventive means against infection-mediated neurodevelopmental disorders. The continuous implementation and integration of preclinical studies and human epidemiological research will be pivotal in overcoming these hurdles and may eventually help reducing or even preventing the burdens associated with infection-mediated neurodevelopmental disorders.
References
Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH . Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol 2013; 12: 406–414.
Paus T, Keshavan M, Giedd JN . Why do many psychiatric disorders emerge during adolescence? Nat Rev Neurosci 2008; 9: 947–957.
Silberberg D, Anand NP, Michels K, Kalaria RN . Brain and other nervous system disorders across the lifespan - global challenges and opportunities. Nature 2015; 527: S151–S154.
Harrison PJ, Weinberger DR . Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 2005; 10: 40–68.
Brown AS, Derkits EJ . Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 2010; 167: 261–280.
Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS . Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry 2013; 70: 677–685.
Atladottir HO, Henriksen TB, Schendel DE, Parner ET . Autism after infection, febrile episodes, and antibiotic use during pregnancy: an exploratory study. Pediatrics 2012; 130: e1447–e1454.
Brown AS, Sourander A, Hinkka-Yli-Salomaki S, McKeague IW, Sundvall J, Surcel HM . Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry 2014; 19: 259–264.
Canetta SE, Bao Y, Co MD, Ennis FA, Cruz J, Terajima M et al. Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am J Psychiatry 2014; 171: 557–563.
Harvey L, Boksa P . Prenatal and postnatal animal models of immune activation: relevance to a range of neurodevelopmental disorders. Dev Neurobiol 2012; 72: 1335–1348.
Meyer U . New serological evidence points toward an infectious route to bipolar disorder. Am J Psychiatry 2014; 171: 485–488.
Owen MJ . Intellectual disability and major psychiatric disorders: a continuum of neurodevelopmental causality. Br J Psychiatry 2012; 200: 268–269.
Meyer U . Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry 2014; 75: 307–315.
Meyer U, Feldon J, Schedlowski M, Yee BK . Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev 2005; 29: 913–947.
Meyer U, Feldon J . Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol 2010; 90: 285–326.
Meyer U, Feldon J, Fatemi SH . In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev 2009; 33: 1061–1079.
Estes ML, McAllister AK . Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016; 353: 772–777.
Bauman MD, Iosif AM, Smith SE, Bregere C, Amaral DG, Patterson PH . Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol Psychiatry 2014; 75: 332–341.
Weir RK, Forghany R, Smith SE, Patterson PH, McAllister AK, Schumann CM et al. Preliminary evidence of neuropathology in nonhuman primates prenatally exposed to maternal immune activation. Brain Behav Immun 2015; 48: 139–146.
Machado CJ, Whitaker AM, Smith SE, Patterson PH, Bauman MD . Maternal immune activation in nonhuman primates alters social attention in juvenile offspring. Biol Psychiatry 2015; 77: 823–832.
Bohacek J, Mansuy IM . Molecular insights into transgenerational non-genetic inheritance of acquired behaviours. Nat Rev Genet 2015; 16: 641–652.
Szyf M . Prospects for the development of epigenetic drugs for CNS conditions. Nat Rev Drug Discov 2015; 14: 461–474.
Silasi M, Cardenas I, Kwon JY, Racicot K, Aldo P, Mor G . Viral infections during pregnancy. Am J Reprod Immunol 2015; 73: 199–213.
Yazigi A, De Pecoulas AE, Vauloup-Fellous C, Grangeot-Keros L, Ayoubi JM, Picone O . Fetal and neonatal abnormalities due to congenital rubella syndrome: a review of literature. J Matern Fetal Neonatal Med 2016; 1–5.
Smith SE, Li J, Garbett K, Mirnics K, Patterson PH . Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 2007; 27: 10695–10702.
Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016; 351: 933–939.
Girard S, Tremblay L, Lepage M, Sebire G . IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J Immunol 2010; 184: 3997–4005.
Traynor TR, Majde JA, Bohnet SG, Krueger JM . Intratracheal double-stranded RNA plus interferon-gamma: a model for analysis of the acute phase response to respiratory viral infections. Life Sci 2004; 74: 2563–2576.
Triantafilou M, Brandenburg K, Gutsmann T, Seydel U, Triantafilou K . Innate recognition of bacteria: engagement of multiple receptors. Crit Rev Immunol 2002; 22: 251–268.
Dahlgren J, Samuelsson AM, Jansson T, Holmang A . Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res 2006; 60: 147–151.
Zaretsky MV, Alexander JM, Byrd W, Bawdon RE . Transfer of inflammatory cytokines across the placenta. Obstet Gynecol 2004; 103: 546–550.
Holmlund U, Cebers G, Dahlfors AR, Sandstedt B, Bremme K, Ekstrom ES et al. Expression and regulation of the pattern recognition receptors Toll-like receptor-2 and Toll-like receptor-4 in the human placenta. Immunology 2002; 107: 145–151.
Schaefer TM, Fahey JV, Wright JA, Wira CR . Innate immunity in the human female reproductive tract: antiviral response of uterine epithelial cells to the TLR3 agonist poly(I:C). J Immunol 2005; 174: 992–1002.
Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I et al. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci 2006; 26: 4752–4762.
Nathan C, Shiloh MU . Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA 2000; 97: 8841–8848.
Lante F, Meunier J, Guiramand J, Maurice T, Cavalier M, de Jesus Ferreira MC et al. Neurodevelopmental damage after prenatal infection: role of oxidative stress in the fetal brain. Free Radic Biol Med 2007; 42: 1231–1245.
Dantzer R . Cytokine, sickness behavior, and depression. Immunol Allergy Clin North Am 2009; 29: 247–264.
Lakshmy R . Metabolic syndrome: role of maternal undernutrition and fetal programming. Rev Endocr Metab Disord 2013; 14: 229–240.
Marques AH, Bjorke-Monsen AL, Teixeira AL, Silverman MN . Maternal stress, nutrition and physical activity: Impact on immune function, CNS development and psychopathology. Brain Res 2015; 1617: 28–46.
Aguilar-Valles A, Flores C, Luheshi GN . Prenatal inflammation-induced hypoferremia alters dopamine function in the adult offspring in rat: relevance for schizophrenia. PLoS ONE 2010; 5: e10967.
Kwik-Uribe CL, Gietzen D, German JB, Golub MS, Keen CL . Chronic marginal iron intakes during early development in mice result in persistent changes in dopamine metabolism and myelin composition. J Nutr 2000; 130: 2821–2830.
Hsiao EY, Patterson PH . Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun 2011; 25: 604–615.
Carpentier PA, Dingman AL, Palmer TD . Placental TNF-alpha signaling in illness-induced complications of pregnancy. Am J Pathol 2011; 178: 2802–2810.
Ribeiro CF, Lopes VG, Brasil P, Pires AR, Rohloff R, Nogueira RM . Dengue infection in pregnancy and its impact on the placenta. Int J Infect Dis 2017; 55: 109–112.
Bale TL . Epigenetic and transgenerational reprogramming of brain development. Nat Rev Neurosci 2015; 16: 332–344.
Sato F, Tsuchiya S, Meltzer SJ, Shimizu K . MicroRNAs and epigenetics. FEBS J 2011; 278: 1598–1609.
Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 2011; 478: 519–523.
Hirabayashi Y, Gotoh Y . Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci 2010; 11: 377–388.
Spiers H, Hannon E, Schalkwyk LC, Smith R, Wong CC, O'Donovan MC et al. Methylomic trajectories across human fetal brain development. Genome Res 2015; 25: 338–352.
Schneider E, Dittrich M, Bock J, Nanda I, Muller T, Seidmann L et al. CpG sites with continuously increasing or decreasing methylation from early to late human fetal brain development. Gene 2016; 592: 110–118.
Feng J, Chang H, Li E, Fan G . Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J Neurosci Res 2005; 79: 734–746.
Guy J, Cheval H, Selfridge J, Bird A . The role of MeCP2 in the brain. Annu Rev Cell Dev Biol 2011; 27: 631–652.
Jakovcevski M, Akbarian S . Epigenetic mechanisms in neurological disease. Nat Med 2012; 18: 1194–1204.
Feng J, Fan G . The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int Rev Neurobiol 2009; 89: 67–84.
Hutnick LK, Golshani P, Namihira M, Xue Z, Matynia A, Yang XW et al. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet 2009; 18: 2875–2888.
Kondo T, Raff M . Chromatin remodeling and histone modification in the conversion of oligodendrocyte precursors to neural stem cells. Genes Dev 2004; 18: 2963–2972.
Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH . Recovery of learning and memory is associated with chromatin remodelling. Nature 2007; 447: 178–182.
Kim J, Krichevsky A, Grad Y, Hayes GD, Kosik KS, Church GM et al. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc Natl Acad Sci USA 2004; 101: 360–365.
Ponomarev ED, Veremeyko T, Barteneva NS . Visualization and quantitation of the expression of microRNAs and their target genes in neuroblastoma single cells using imaging cytometry. BMC Res Notes 2011; 4: 517.
Moreau MP, Bruse SE, Jornsten R, Liu Y, Brzustowicz LM . Chronological changes in microRNA expression in the developing human brain. PLoS ONE 2013; 8: e60480.
Gapp K, von Ziegler L, Tweedie-Cullen RY, Mansuy IM . Early life epigenetic programming and transmission of stress-induced traits in mammals: how and when can environmental factors influence traits and their transgenerational inheritance? Bioessays 2014; 36: 491–502.
Franklin TB, Russig H, Weiss IC, Graff J, Linder N, Michalon A et al. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry 2010; 68: 408–415.
Gapp K, Jawaid A, Sarkies P, Bohacek J, Pelczar P, Prados J et al. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat Neurosci 2014; 17: 667–669.
Bohacek J, Farinelli M, Mirante O, Steiner G, Gapp K, Coiret G et al. Pathological brain plasticity and cognition in the offspring of males subjected to postnatal traumatic stress. Mol Psychiatry 2015; 20: 621–631.
Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL . Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci 2013; 33: 9003–9012.
Rodgers AB, Morgan CP, Leu NA, Bale TL . Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc Natl Acad Sci USA 2015; 112: 13699–13704.
Morgan CP, Bale TL . Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J Neurosci 2011; 31: 11748–11755.
Yao Y, Robinson AM, Zucchi FC, Robbins JC, Babenko O, Kovalchuk O et al. Ancestral exposure to stress epigenetically programs preterm birth risk and adverse maternal and newborn outcomes. BMC Med 2014; 12: 121.
Jensen Pena C, Monk C, Champagne FA . Epigenetic effects of prenatal stress on 11beta-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS ONE 2012; 7: e39791.
Veenendaal MV, Painter RC, de Rooij SR, Bossuyt PM, van der Post JA, Gluckman PD et al. Transgenerational effects of prenatal exposure to the 1944-45 Dutch famine. BJOG 2013; 120: 548–553.
Dunn GA, Bale TL . Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 2011; 152: 2228–2236.
Rechavi O, Houri-Ze'evi L, Anava S, Goh WS, Kerk SY, Hannon GJ et al. Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell 2014; 158: 277–287.
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA 2008; 105: 17046–17049.
Zheng S, Rollet M, Yang K, Pan YX . A gestational low-protein diet represses p21(WAF1/Cip1) expression in the mammary gland of offspring rats through promoter histone modifications. Br J Nutr 2012; 108: 998–1007.
Masuyama H, Mitsui T, Nobumoto E, Hiramatsu Y . The effects of high-fat diet exposure in utero on the obesogenic and diabetogenic traits through epigenetic changes in adiponectin and leptin gene expression for multiple generations in female mice. Endocrinology 2015; 156: 2482–2491.
Anway MD, Cupp AS, Uzumcu M, Skinner MK . Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005; 308: 1466–1469.
Skinner MK, Anway MD, Savenkova MI, Gore AC, Crews D . Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS ONE 2008; 3: e3745.
Stouder C, Paoloni-Giacobino A . Transgenerational effects of the endocrine disruptor vinclozolin on the methylation pattern of imprinted genes in the mouse sperm. Reproduction 2010; 139: 373–379.
Faulk C, Kim JH, Anderson OS, Nahar MS, Jones TR, Sartor MA et al. Detection of differential DNA methylation in repetitive DNA of mice and humans perinatally exposed to bisphenol A. Epigenetics 2016; 11: 489–500.
Wolstenholme JT, Edwards M, Shetty SR, Gatewood JD, Taylor JA, Rissman EF et al. Gestational exposure to bisphenol a produces transgenerational changes in behaviors and gene expression. Endocrinology 2012; 153: 3828–3838.
Guerrero-Bosagna C, Covert TR, Haque MM, Settles M, Nilsson EE, Anway MD et al. Epigenetic transgenerational inheritance of vinclozolin induced mouse adult onset disease and associated sperm epigenome biomarkers. Reprod Toxicol 2012; 34: 694–707.
Vassoler FM, White SL, Schmidt HD, Sadri-Vakili G, Pierce RC . Epigenetic inheritance of a cocaine-resistance phenotype. Nat Neurosci 2013; 16: 42–47.
Govorko D, Bekdash RA, Zhang C, Sarkar DK . Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol Psychiatry 2012; 72: 378–388.
Meyer U, Feldon J . To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology 2012; 62: 1308–1321.
Nilsson EE, Skinner MK . Environmentally induced epigenetic transgenerational inheritance of disease susceptibility. Transl Res 2015; 165: 12–17.
Hollins SL, Zavitsanou K, Walker FR, Cairns MJ . Alteration of imprinted Dlk1-Dio3 miRNA cluster expression in the entorhinal cortex induced by maternal immune activation and adolescent cannabinoid exposure. Transl Psychiatry 2014; 4: e452.
Tang B, Jia H, Kast RJ, Thomas EA . Epigenetic changes at gene promoters in response to immune activation in utero. Brain Behav Immun 2013; 30: 168–175.
Basil P, Li Q, Dempster EL, Mill J, Sham PC, Wong CC et al. Prenatal maternal immune activation causes epigenetic differences in adolescent mouse brain. Transl Psychiatry 2014; 4: e434.
Labouesse MA, Dong E, Grayson DR, Guidotti A, Meyer U . Maternal immune activation induces GAD1 and GAD2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 2015; 10: 1143–1155.
Richetto J, Massart R, Weber-Stadlbauer U, Szyf M, Riva M, Meyer U . Genome-wide DNA methylation changes in a mouse model of infection-mediated neurodevelopmental disorders. Biol Psychiatry 2016; 81: 265–276.
Ronovsky M, Berger S, Zambon A, Reisinger SN, Horvath O, Pollak A et al. Maternal immune activation transgenerationally modulates maternal care and offspring depression-like behavior. Brain Behav Immun 2016; pii: S0889-1591(16)30480-9.
Weber-Stadlbauer U, Richetto J, Labouesse MA, Bohacek J, Mansuy IM, Meyer U . Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol Psychiatry 2016; 22: 102–112.
Apostolou E, Hochedlinger K . Chromatin dynamics during cellular reprogramming. Nature 2013; 502: 462–471.
Meyer U, Feldon J, Yee BK . A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr Bull 2009; 35: 959–972.
Hodge DR, Cho E, Copeland TD, Guszczynski T, Yang E, Seth AK et al. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genomics Proteomics 2007; 4: 387–398.
Foran E, Garrity-Park MM, Mureau C, Newell J, Smyrk TC, Limburg PJ et al. Upregulation of DNA methyltransferase-mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol Cancer Res 2010; 8: 471–481.
Zijlstra GJ, Ten Hacken NH, Hoffmann RF, van Oosterhout AJ, Heijink IH . Interleukin-17 A induces glucocorticoid insensitivity in human bronchial epithelial cells. Eur Respir J 2012; 39: 439–445.
Rahman I, Gilmour PS, Jimenez LA, MacNee W . Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem 2002; 234-235: 239–248.
Ausio J . MeCP2 and the enigmatic organization of brain chromatin. Implications for depression and cocaine addiction. Clin Epigenetics 2016; 8: 58.
Wang G, Dai J, Mao J, Zeng X, Yang X, Wang X . Folic acid reverses hyper-responsiveness of LPS-induced chemokine secretion from monocytes in patients with hyperhomocysteinemia. Atherosclerosis 2005; 179: 395–402.
Kolb AF, Petrie L . Folate deficiency enhances the inflammatory response of macrophages. Mol Immunol 2013; 54: 164–172.
Oakley GP Jr, Erickson JD, James LM, Mulinare J, Cordero JF . Prevention of folic acid-preventable spina bifida and anencephaly. Ciba Found Symp 1994; 181: 212–223, discussion 23-31.
Gao Y, Sheng C, Xie RH, Sun W, Asztalos E, Moddemann D et al. New perspective on impact of folic acid supplementation during pregnancy on neurodevelopment/autism in the offspring children - a systematic review. PLoS ONE 2016; 11: e0165626.
Brown AS . The environment and susceptibility to schizophrenia. Prog Neurobiol 2011; 93: 23–58.
Cao B, Wang DF, Xu MY, Liu YQ, Yan LL, Wang JY et al. Lower folate levels in schizophrenia: a meta-analysis. Psychiatry Res 2016; 245: 1–7.
Ramaekers VT, Blau N, Sequeira JM, Nassogne MC, Quadros EV . Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics 2007; 38: 276–281.
Schmidt RJ, Tancredi DJ, Ozonoff S, Hansen RL, Hartiala J, Allayee H et al. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study. Am J Clin Nutr 2012; 96: 80–89.
Barua S, Kuizon S, Junaid MA . Folic acid supplementation in pregnancy and implications in health and disease. J Biomed Sci 2014; 21: 77.
Niculescu MD, Zeisel SH . Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J Nutr 2002; 132: 2333S–2335SS.
Joubert BR, den Dekker HT, Felix JF, Bohlin J, Ligthart S, Beckett E et al. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat Commun 2016; 7: 10577.
Sender R, Fuchs S, Milo R . Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 2016; 14: e1002533.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS et al. Evolution of mammals and their gut microbes. Science 2008; 320: 1647–1651.
Cryan JF, Dinan TG . Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 2012; 13: 701–712.
Jimenez E, Marin ML, Martin R, Odriozola JM, Olivares M, Xaus J et al. Is meconium from healthy newborns actually sterile? Res Microbiol 2008; 159: 187–193.
Wassenaar TM, Panigrahi P . Is a foetus developing in a sterile environment? Lett Appl Microbiol 2014; 59: 572–579.
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155: 1451–1463.
Kratsman N, Getselter D, Elliott E . Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 2016; 102: 136–145.
Davie JR . Inhibition of histone deacetylase activity by butyrate. J Nutr 2003; 133: 2485 S–2493.
Meyer U, Nyffeler M, Schwendener S, Knuesel I, Yee BK, Feldon J . Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology 2008; 33: 441–456.
Fortier ME, Luheshi GN, Boksa P . Effects of prenatal infection on prepulse inhibition in the rat depend on the nature of the infectious agent and the stage of pregnancy. Behav Brain Res 2007; 181: 270–277.
Kohli RM, Zhang Y . TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013; 502: 472–479.
Seisenberger S, Peat JR, Hore TA, Santos F, Dean W, Reik W . Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philos Trans R Soc Lond B Biol Sci 2013; 368: 20110330.
Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR . Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med 2016; 374: 1981–1987.
Zhubi A, Cook EH, Guidotti A, Grayson DR . Epigenetic mechanisms in autism spectrum disorder. Int Rev Neurobiol 2014; 115: 203–244.
Costa E, Dong E, Grayson DR, Ruzicka WB, Simonini MV, Veldic M et al. Epigenetic targets in GABAergic neurons to treat schizophrenia. Adv Pharmacol 2006; 54: 95–117.
Guidotti A, Auta J, Chen Y, Davis JM, Dong E, Gavin DP et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 2011; 60: 1007–1016.
Nestler EJ, Pena CJ, Kundakovic M, Mitchell A, Akbarian S . Epigenetic basis of mental illness. Neuroscientist 2015; 22: 447–463.
Gavin DP, Chase KA, Sharma RP . Active DNA demethylation in post-mitotic neurons: a reason for optimism. Neuropharmacology 2013; 75: 233–245.
Dube E, Vivion M, MacDonald NE . Vaccine hesitancy, vaccine refusal and the anti-vaccine movement: influence, impact and implications. Expert Rev Vaccines 2015; 14: 99–117.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Weber-Stadlbauer, U. Epigenetic and transgenerational mechanisms in infection-mediated neurodevelopmental disorders. Transl Psychiatry 7, e1113 (2017). https://doi.org/10.1038/tp.2017.78
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2017.78
This article is cited by
-
Brain methylome remodeling selectively regulates neuronal activity genes linking to emotional behaviors in mice exposed to maternal immune activation
Nature Communications (2023)
-
Post-stroke depression: epigenetic and epitranscriptomic modifications and their interplay with gut microbiota
Molecular Psychiatry (2023)
-
Behavioral, neuroanatomical, and molecular correlates of resilience and susceptibility to maternal immune activation
Molecular Psychiatry (2021)
-
Maternal immune activation and neuroinflammation in human neurodevelopmental disorders
Nature Reviews Neurology (2021)
-
Exploring the evidence for epigenetic regulation of environmental influences on child health across generations
Communications Biology (2021)
